
Abstract
Radioactivity has been monitored in seafloor sediments off Fukushima and nearby prefectures regularly. During the initial monitoring period (May–September 2011), 137Cs concentrations in the surface sediments (0–3 cm) generally increased to 8–580 Bq/kg. Subsequently, concentrations decreased at variable rates. In the latest data, from February 2016, concentrations were still higher at 0.8–141 Bq/kg than the pre-accident level. The geometric mean concentration declined steadily from 47 Bq/kg in September 2011 to 13 Bq/kg in February 2016. The 137Cs abundance (Bq/m2) in the surface sediment at each station decreased similarly. The rate of decrease of surface abundance varied spatially by almost one order of magnitude, ranging from 1.1 × 10−4 to 1.7 × 10−3/day, equivalent to halving times of 16–1.1 years, respectively. The rate of decrease was related to the median sediment grain size at each station. In addition, bottom-water dynamics, through the redistribution of bottom sediments, may have caused spatial variability in the rate of decrease, whereas vertical profiles of 137Cs concentrations in the sediment suggest that vertical migration of 137Cs was not a major mechanism reducing the surface 137Cs concentration. From September 2011 to February 2016, the overall halving time of 137Cs in the surface sediment in the monitoring area, excluding the area inside a 30-km radius from the Fukushima Dai-ichi Nuclear Power Plant, was 2.3 years. Thus, 76% of the originally deposited 137Cs (46 × 1012 Bq) in the surface sediment was transported out of the area during that period.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The 2011 Tohoku Earthquake off the Pacific coast of Japan occurred on 11 March 2011, and the associated tsunami damaged the Fukushima Daiichi Nuclear Power Plant (FDNPP) of the Tokyo Electric Power Company (TEPCO), resulting in the release of large amounts of radioactive material to the marine environment, including the seawater and bottom sediments. For example, the 137Cs concentration in seawater outside a 30-km radius from the FDNPP rose from a pre-accident level of 0.0011–0.0019 Bq/l to as much as 186 Bq/l in April 2011 (Oikawa et al. 2013). Since then, the concentration has been decreasing as a result of the mixing and migration of waters to levels almost the same as or slightly higher than the pre-accident level (Takata et al. 2016).
Radioactivity in seafloor sediments has been systematically monitored by the Marine Ecology Research Institute, under contract with the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT), from May 2011 to March 2013, and with the Secretariat of the Nuclear Regulation Authority of Japan (NRA) from April 2013 to the present. Initially, there were eight monitoring stations, but the number has increased gradually; since May 2012, 32 monitoring stations have been in operation off Fukushima and nearby prefectures. Kusakabe et al. (2013) have already reported monitoring results obtained during the period from May 2011 to February 2012. In particular, they make the following points:
-
Concentrations of 137Cs in the surface sediments varied spatially by two orders of magnitude, from 1.7 to 580 Bq/kg.
-
There was no obvious correlation between the 137Cs concentration and the proximity of the sampling location to the accident site.
-
The estimated abundance of 137Cs accumulated in the upper 3 cm of surface sediments in the monitoring area was 3.78 × 1013 Bq in May 2011, and it declined subsequently.
-
Spatial variations in the 137Cs concentration and inventory depended on two main factors: the 137Cs concentration in the overlying water during the first several months after the accident and the physical characteristics of the sediments (i.e., water content and bulk density).
Several possible modes of 137Cs deposition and post-depositional migration have been proposed. For example, Misumi et al. (2014) indicated that for the first several months after the accident, the horizontal distribution of 137Cs in the surface sediment was related to its deposition and adsorption from the polluted seawater. Ambe et al. (2014) identified a high-concentration band with a width of about 20 km in the southern part of the monitoring area, where waters were shallower than 100 m, and showed that the higher concentrations were due to adsorption by fine-grained sediment, and Otosaka and Kobayashi (2013) proposed that 137Cs-enriched fine particles were transported offshore. Long-term monitoring of bottom waters just above the seafloor and field experiments carried out off the Fukushima coast revealed that the combination of wave and current action caused by meteorological disturbance was a key process in the transport of suspended particulate material (Yagi et al. 2015; Buesseler et al. 2015). On the basis of a numerical simulation, Higashi et al. (2015) suggested that the shape of the high-concentration band in the southern area (the “hotspot swath”) mainly reflected spatiotemporal variation in bottom shear stress between the shallow shelf (<50 m water depth) and the area offshore of the shelf break. These proposals are all based on data obtained during a relatively restricted time span after the accident (~1 year), so long-term trends of 137Cs deposition and transport in the area are not yet known. Furthermore, the temporal trend of the 137Cs inventory in the monitoring area when the 137Cs profile to a depth of 3 cm or more is taken into account needs to be assessed.
Several studies have investigated the 137Cs budget in the sediment off Fukushima (Ambe et al. 2014; Black and Buesseler 2014; Otosaka and Kato 2014; Higashi et al. 2015), but because the sampling area and timing differed among these studies, it is difficult to compare the data to clarify the fate of FDNPP-derived 137Cs in the sediment. In the Irish Sea, MacKenzie et al. (1994) reported that sediment contaminated by radionuclides from the Sellafield nuclear fuel reprocessing plant is redistributed mainly with silt-sized sediment particles, and dissolution of 137Cs from the sediment may also play an important role in post-depositional migration of 137Cs (e.g., Cook et al. 1997; MacKenzie et al. 1998). A detailed study of the horizontal distribution of FDNPP-derived radionuclides, especially radiocesium, and its temporal change is thus required.
In this study, we analyzed the MEXT and NRA monitoring data on radioactive Cs (134Cs, 137Cs) concentrations in bottom sediments collected from May 2011 to February 2016 and evaluated the radiocesium inventory in the sediments. Then, we examined factors controlling the post-depositional migration of FDNPP-derived radioactive Cs in the monitoring area and the reason for the declining trend of the Cs inventory by considering additional relevant data such as sediment texture.
2 Methods
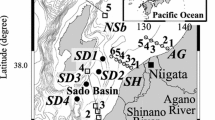
Collection of bottom sediments at a limited number of sampling locations began in May 2011, and since May 2012, monitoring has been conducted four times a year at 32 sampling stations (Fig. 1; Table S1). Note that the area within a 30-km radius from the FDNPP was not surveyed by this study. Bottom sediments were retrieved with a multiple corer equipped with eight plastic tubes (opening 8.2 cm in diameter). The top end of each tube was sealed tightly on retrieval so that the surface sediment was obtained intact. If the surface sediment was disturbed, it was discarded. The upper 3 cm of sediment from the eight sediment cores was combined and analyzed for radiocesium activity and other physical and chemical parameters. Occasionally, more cores from additional casts were used in the analysis, depending on the weight of the sediments obtained during the initial cast. We also studied vertical profiles of 137Cs and 134Cs in sediments in cores obtained at several selected locations. In particular, we studied the vertical profiles of 137Cs in sediment cores collected since May 2012 at nine stations. Because the subsampled layers used for the profile analysis were thinner than those used for the 3-cm surface sediment analyses, more cores needed to be collected to obtain vertical profile samples of adequate size compared with the number needed for the surface sediment analyses; in most cases, subsamples from 24 cores were combined and analyzed.

Locations of sampling stations. The red star and line indicate the location of the Fukushima Dai-ichi Nuclear Power Plant and the 30-km radius, respectively
A portion of the wet sediment of each sample was packed into a 120-ml plastic bottle and saved for later measurement of bulk density and water content by the gravimetric method in the laboratory on land. The remainder of each sample was dried at 105 °C, ground in a mortar, sieved through a screen (mesh size <2 mm), mixed well, and then pulverized to a homogeneous powder in a table-top grinder.
An aliquot of each dried sediment sample (400–600 g) was placed in a plastic container and analyzed by nondestructive gamma-ray spectrometry with a Ge detector. Details of the quantification of 134Cs and 137Cs activities are described in Kusakabe et al. (2013).
Physical characteristics of the surface sediment, in particular, grain size and organic components (loss on ignition, organic carbon, and organic nitrogen), were measured in samples obtained at 13 different collection times from May 2012 to November 2015. The grain-size distribution of the sediment samples was assessed by a sieving analysis (Japanese Industrial Standard 2009). Loss on ignition was measured by calculating the weight difference between before and after heating at 600 °C for 1 h. A CHN analyzer was used to measure the organic carbon and nitrogen contents of the sediment.
3 Results and discussion
The concentration data for 137Cs and 134Cs in the surface sediments were published on the NRA website (http://radioactivity.nsr.go.jp/ja/list/458/list-1.html) as soon as they became available. These radionuclide data, together with all other relevant data used in this report, are summarized in Supplementary Tables S1–S4.
3.1 Sedimentary setting of the monitoring area
Most sampling stations in the monitoring area are located on the continental shelf to slope at water depths ranging from ~30 m (Stn. K1) to ~670 m (Stn. G4) (Fig. 1). Sediments in the study area had a wide variety of textures and chemical characteristics (Tables 1 and S3; Fig. 2). Sediment samples from Stns. B1, L1, and C1 were clearly distinguishable from the rest of the samples by their grain size distributions and median grain size (D50); that is, these samples were high in medium and coarse sand and had a larger median grain size, 0.75–0.88 mm, on average. In addition, the median grain size at Stn. J1 was relatively high on average, and its temporal variation, from 0.17 to 1.1 mm, was quite large compared with that at the other stations (Table S3); this variation may reflect fine spatial variation of texture at Stn. J1 or, given the shallow water depth of the station, rapid temporal changes due to agitation by waves and tidal currents in the overlying seawater. Moreover, fluvial input of particles from nearby rivers (i.e., Kuji River and Naka River) may have some impact on the distribution of particles and its temporal change in the area (Takata et al. 2015). These grain size characteristics may affect the temporal variation of the 137Cs concentration in the sediment at this station (discussed in Sect. 3.5).

Texture of the sediments. The data shown are the averages of samples collected from May 2012 to November 2014. See Table S2 for details. Upper panel grain size distributions. Because the sediments from Stns. B1, L1, C1, J1, K1, and M1 contained particles larger than fine pebbles, their percentages sum to less than 100%. Lower panel median grain size at each station
Although physical and chemical parameters were quite variable among the stations (Fig. 2; Table S3), some parameters co-varied (Table 2). Parameters related to the physical texture, such as median grain size and silt and clay contents, were closely correlated with biogenic parameters such as ignition loss and organic carbon and nitrogen contents. Interestingly, silt and clay contents were also correlated with biogenic parameters. For example, the correlation coefficient (r) between silt and clay contents and ignition loss was about 0.8. The surface distribution of silt and clay contents averaged over 11 observations from May 2012 to November 2014 (Fig. 3) shows that, generally, sediments rich in fine particles were dominant in the southern part of the monitoring area, namely, at Stns. I0, I1, and J3, and to a lesser extent at E1, F1, and G0. At one northern station, B3, the silt and clay contents were substantially higher than at other northern stations. This result is consistent with the detailed studies reported by Ambe et al. (2014).

Spatial variation of silt + clay (grain size <0.075 mm) contents. The data represent average values during May 2012–November 2014
3.2 Distributions of 137Cs concentrations in surface sediment
We examined temporal changes of the 137Cs concentrations in the surface sediment to a depth of 3 cm in this study as well as the changes during the initial monitoring period (May 2011–September 2011) reported previously (Kusakabe et al. 2013) (Fig. 4). During the initial monitoring period, 137Cs concentrations generally increased with time to a variable degree among the stations, reaching maximum values of up to 520 (Stn. J1) Bq/kg in dry weight (Kusakabe et al. 2013), and they subsequently decreased. In February 2016, they ranged from 0.8 Bq/kg at Stn. L1 to 141 Bq/kg, at Stn. I1. In general, the geometric mean concentration decreased steadily from 47 Bq/kg in September 2011 to 13 Bq/kg in February 2016, though the concentration at individual stations showed sporadic peaks. An exponential function fitted to the geometric means gives an apparent halving time of 2.26 years. Despite the decreasing trend in the monitoring area, in February 2016, all values still exceeded the pre-accident 5-year average level (2006–2010) at eight stations off Fukushima Prefecture (0.87 ± 0.41 Bq/kg; Kusakabe et al. 2013), though not in samples collected near the coast in the southernmost part of the sampling area (Stns. K1, L1 and M1). This southernmost area is influenced by the Kuroshio current, which originates from a remote area with minimal amounts of FDNPP-derived 137Cs, and the sediments at those stations also have a relatively higher sand content (Figs. 2, 3), a reflection of the existence of an intense hydrodynamic current in that area.

Temporal changes in surface 137Cs concentrations. Some data are from Kusakabe et al. (2013). The thick black line represents the geometric mean of the concentrations obtained at all stations during the same cruise. The mean is not shown before September 2011 because the number of sampling stations was much smaller before that date
The declining trend of 137Cs with time in the surface sediments was spatially variable (Fig. 5). From September 2011 to February 2012, relatively higher concentrations were found in two distinctive areas, one in the northern (e.g., Stns. D1 and B3) and the other in the southern (e.g., Stn. J1) part of the monitoring area. Although the concentration decreased gradually at most stations, at Stns. I0 and I1 137Cs concentrations decreased less than at other stations, and they were the highest among all stations in February 2016; the halving times of surface 137Cs concentrations at Stns. I0 and I1 were calculated to be 3.07 and 5.46 years, respectively. In addition, the whole-core inventory at Stn. I1 decreased to a lesser extent with time compared to the rest of the samples (see Sect. 3.6.2).

Spatial variations and temporal changes in the surface 137Cs concentration. Some data are from Kusakabe et al. (2013)
Interestingly, the 137Cs concentration does not necessarily reflect the proximity of the sampling station to the FDNPP. Kusakabe et al. (2013) found that the average concentration in the surface sediment was related to both the integrated concentration in the overlying seawater column and the sediment bulk density (which correlates with grain size). Because not many measurements of 137Cs concentrations in seawater were obtained in the first several months after the accident, the findings of Kusakabe et al. (2013) imply that the pathway of the polluted seawater along currents after the accident may be one of the main factors determining the initial surface distribution.
In many cases, the deviation of the 137Cs concentration in the sediments at each sampling station from the temporal geometric mean was greater than the pre-accident concentration variation (ca. 50%). The 137Cs concentration has been reported to vary spatially within a small area. For example, 137Cs concentrations of six sediment sampling casts performed at the same station (Stn. D1) in a single day showed wide variability (170–580 Bq/kg), even though for each cast the concentration was measured after combining the sediments from the eight individual cores (Kusakabe et al. 2013). Thornton et al. (2013a), who developed a gamma ray spectrometer that measures in situ radionuclide distributions in the surface sediment with high spatial resolution (<10 m), reported high concentration anomalies over distances from a few meters to a few hundred meters and demonstrated that the distribution of the anomalies was associated with meter-scale features of the bottom topography. So-called Cs balls, which are particles with a diameter of a few micrometers containing extremely high 137Cs concentrations (0.66–3.27 Bq/particle), were released into the air by the accident (Adachi et al. 2013). Recently, Ikenoue et al. (2017) have hypothesized that consistently high but variable concentrations of 137Cs in zooplankton collected from the waters off Fukushima and nearby prefectures, compared with concentrations in the ambient seawater, are mainly attributable to the presence of 137Cs-enriched particles. Although Cs balls or 137Cs-enriched particles have not yet been reported in marine sediments, it is possible that the concentration variability in sediments might be at least partially due to the presence of such particles. The high temporal variability of the 137Cs concentrations at each station (Fig. 4), despite the overall declining trend, may thus reflect spatial variations within a relatively small area related to bottom topography and the presence of particles containing extremely high 137Cs concentrations.
3.3 Distributions of 134Cs concentrations in surface sediment
The distribution of 134Cs in the surface sediments should mimic that of 137Cs because the initial 134Cs/137Cs ratio at the time of the accident was 1 (e.g., Aoyama et al. 2012; Buesseler et al. 2012). Differences in the distributions of the two isotopes arise because of the shorter half-life of 134Cs and the presence of 137Cs fallout. Temporally, the 134Cs/137Cs ratios decreased with time in parallel with the radioactive decay curve (Fig. 6, black line), which takes into account the decays of both 134Cs and 137Cs, but in general the data points were below the curve owing to the influence of global fallout. In particular, data from sediments with relatively low concentrations of FDNPP-derived 137Cs, such as the data from Stns. A1, B5, E5, K1, L1, and M1, deviated from the line; above all, the data from Stn. M1 showed the largest deviation, reflecting the fact that this station, the southernmost (Fig. 1), was least affected by the accident, as described in Sect. 3.2. The 134Cs and 137Cs concentrations in the bottom water (i.e., about 100 m above the seafloor) were also measured during the NRA monitoring project. Interestingly, the bottom water 134Cs/137Cs ratios at the southern stations (e.g., K1, L1, L3, and M1) showed the largest deviations from the theoretical decay curve among the stations (Takata et al. 2016). This result implies that relatively less polluted water intruded from outside the monitoring area, most likely from the south. Nevertheless, the 134Cs/137Cs ratio in sediment showed a notably large deviation from the decay curve only at Stn. M1, and it approached the curve over time. This result suggests that the 137Cs in the sediment right after the accident was dominated by 137Cs of fallout origin with little 134Cs and that Stn. M1 subsequently received additional 134Cs and 137Cs inputs that compensated for the decay of 134Cs, so that the ratio eventually converged to the calculated decay curve. Such additional inputs might have been derived from allochthonous particulate matter with a higher 134Cs/137Cs ratio, probably from the north, where the sediments were more polluted. The delayed arrival of 134Cs (as well as 137Cs) at Stn. M1 may be circumstantial evidence for the transport of radiocesium by particles, because the lateral transport of 134Cs in seawater would be much faster than the lateral transport of particulate matter, which likely would involve repeated cycles of suspension, deposition, and resuspension.

Temporal change of the 134Cs/137Cs ratio in surface sediments. Some data are from Kusakabe et al. (2013). The black line represents the theoretical decay curve, under the assumption of an initial ratio of 1 on 11 March 2011. Colored symbols show data that deviate from the curve seemingly in the figure; other data are shown by thin brown lines
3.4 Vertical distributions of 134Cs and 137Cs
We studied the vertical distribution of 137Cs from May 2012 to October 2015 at nine stations (Fig. 7; Table S4). Although the surface concentration of 137Cs varied by two orders of magnitude among the sampling sites, the concentration profiles were similar, except at Stn. J1; that is, the concentrations decreased downward below the surface mixed layer or subsurface vertical maximum. At Stn. J1, the sand content, which was the highest among the nine stations (Fig. 2), prevented the corer from retrieving cores long enough to observe the overall vertical 137Cs concentration profile. In the cores from Stns. B3, E1, G4, I1, and J3, 137Cs was present at a depth of 20 cm, and 134Cs was also detected at that depth, except at Stn. J3 (Table S4).

Vertical profiles of 137Cs concentrations in the sediments at nine stations. Note that the scale of the horizontal axis differs, depending on the concentration range in each core
Just as the surface 137Cs concentration decreased with time, the concentrations in intermediate and deeper layers also tended to decline, but to a variable extent. The penetration depth of 137Cs in the sediments, however, generally remained almost the same during the sampling period (May 2012–October 2015). Without considering lateral transport of resuspended sediment, Black and Buesseler (2014) estimated the halving time of surface 137Cs due to vertical mixing to be 0.4–26 years. If vertical mixing and downward diffusion were the dominant mechanisms controlling the decrease in the surface concentration, then the Cs concentration in the intermediate and deeper layers, as well as downcore penetration of Cs, should increase with time, but our results show no such increase. The fact that the whole-core inventory of 137Cs also decreased with time in most core samples (see Sect. 3.6.2) suggests that the effect of vertical migration of Cs on the vertical Cs profile was minimal. Only at Stn. E1 did the 137Cs concentration show an obvious increase in intermediate or deeper layers (Fig. 7). Sediment mixing by burrowing animals (Seike et al. 2016) may be responsible for this increase, but quantitative clarification of the role of biological activity in the temporal change of 137Cs distributions requires further study.
3.5 Spatiotemporal change of surface 137Cs abundance
The declining trend of the surface 137Cs concentration (and its abundance) at each station varied spatially. To compare secondary remobilization during the last 5 years (May 2011–February 2016), surface abundance in May 2012, when data became available from all 32 sampling stations, was normalized to 1. Then, we plotted the normalized abundances at each station, together with median grain size data, against time (Fig. 8). During the initial monitoring period (May 2011–August 2011), the abundances increased with time, peaking at different times depending on the station, and then decreased with time to the end of the monitoring period; this pattern was similar to the pattern of the temporal concentration changes (Fig. 4). The rate of decrease varied considerably among the stations. At stations C1, B1, and L1, for example, surface abundance decreased by a factor of tens from May 2011 to February 2016. In general, the rate of decrease appeared to be related to the sediment grain size; during the initial monitoring period, larger median grain sizes (D50 >0.4 mm) were evenly distributed over a wide range of normalized abundances (left side of Fig. 8). Subsequently, 137Cs abundances decreased more steeply in sediment characterized by larger D50 values. This result implies that the main mechanism lowering the 137Cs surface abundance was transport by relatively small, resuspended particles. This mechanism is especially valid for sediments such as those at C1, L1, B1, and J1, which were characterized by a high sand content and large D50 values (Fig. 2; Table 1). It is intuitively reasonable to expect smaller resuspended sediment particles to be more mobile, but this expectation was not necessarily met at all stations (Fig. 8). We therefore examined the relationship between D50 and the rate of decrease further.

Temporal changes in normalized surface abundances. The average median grain size of the sediments is superimposed on the normalized surface abundance at each sampling time. The samples mentioned in the text have colored lines
We determined the rate of decrease at each station starting from September 2011 or May 2012, depending on when data were available, by fitting an exponential function to the abundance data. Because the data were highly variable temporally, for the reasons mentioned in Sect. 3.2, we eliminated data that deviated from the fitted regression line by more than 50% and then fitted the function again to the remaining data to determine the rate of decrease and the correlation coefficient of the regression line. We used a cutoff of 50% because the average 137Cs concentration in the sediment off Fukushima during the 5 years before the accident varied by about 50% (Kusakabe et al. 2013). The calculated rates of decrease ranged from 1.1 × 10−4/day (Stn. IB2) to 1.7 × 10−3/day (Stn. L1); these values are equivalent to halving times of 16 and 1.1 years, respectively (Fig. 9). The correlation coefficients (r) for the regression curves (Fig. 9, color scale) show that at stations where the rate of decrease was smaller than 5 × 10−4/day, the correlation was poor (r < 0.5). This result suggests that the abundance of 137Cs at such stations varied but did not decrease consistently, probably owing to sporadically occurring cycles of deposition and removal of 137Cs in the area. Interestingly, these stations were mostly in the southern part of the 100–200 m isobath zone (except for Stn. B5). If decreases and increases of 137Cs in the surface sediment were controlled dominantly by desorption and adsorption of dissolved Cs in seawater, highly variable temporal changes would not have been observed because the 137Cs concentration in the bottom water did not change significantly over at least the last 3 years (2012–2015) (Takata et al. 2016). Therefore, we inferred that resuspension and deposition were the main mechanisms responsible for the redistribution of surface 137Cs.

Relationship between the rate of decrease of 137Cs surface abundance and median grain size. The color scale shows the correlation coefficient (r) of the regression curve used to derive the rate of decrease
A puzzling feature of Stn. B3 is that, although its sediment D50 value was one of the smallest (Table 1; Fig. 2), its 137Cs removal rate was among the highest (Fig. 9). If vigorous resuspension and lateral transport of sediment had accelerated the surface removal there, then the D50 value should have been higher, absent the persistent addition of less-contaminated small particles. Trawl fishing (e.g., Huang 1982) may have caused resuspension of bottom sediments and led to lateral transport of sedimentary 137Cs. However, trawling has been conducted not only in this area but in other areas as well, so such an artifact may be least likely to be the main cause for the steep decline of 137Cs abundance. The situation at Stn. B3 with respect to 137Cs removal thus needs to be studied further.
Stn. B5 is located outside of Sendai Bay, near where tsunami deposits (Ikehara et al. 2014) and turbidity (Noguchi et al. 2012) resulting from the 2011 earthquake were observed. That sedimentary environment may account for the relatively low and highly variable removal rate of 137Cs at Stn. B5.
3.6 Abundance of 137Cs
3.6.1 Abundance of 137Cs in surface sediments
Kusakabe et al. (2013) calculated the abundance of 137Cs in the surface sediments of the study area from the average of the measured abundances at the stations and the area covered by the stations (22,177 km2). However, the sampling locations were not distributed evenly; because they were more densely distributed in the area closer to the FDNPP, the abundance calculated by Kusakabe et al. (2013) was somewhat biased by samples with higher concentrations collected in a relatively small area. Thus, we re-evaluated the abundance using a different approach and additional data as follows. The region of interest for the calculation, within which all of the sampling stations are located, is bounded by 38°40′N, 35°20′N, and the 800-m depth contour. First, we excluded the area within a radius of 30 km from the FDNPP from the calculation. Then, we partitioned the remainder of the study region into Voronoi polygons, with the boundaries of each polygon drawn such that they were equidistant between neighboring sampling stations (Fig. 10, inset). The total area, that is, the sum of the areas of the Voronoi polygons, was 25,171 km2.

Temporal changes in 137Cs abundance in surface 3 cm of the sediments in the monitoring area. See the text for details of the calculation. T 1/2 represents the halving time of the surface 137Cs abundance. The inset map shows the polygons used to calculate the abundance
We calculated the total 137Cs abundance in surface sediment in the study area by a summation of the abundance in each polygon, which was determined from the area of the polygon containing each station and the abundance at that station (Fig. 10). The total amount of 137Cs in the surface 3 cm of sediment was calculated to be 4.57 × 1013 Bq in September 2011; it decreased with time to 1.09 × 1013 Bq by February 2016. The abundance of 137Cs in the surface sediment in the monitoring area was thus reduced by 76% in 4.5 years. As we stated in Sect. 3.5, surface 137Cs concentrations peaked around the fall of 2011; therefore, we regarded the calculated abundance in September 2011 as the highest abundance ever in the monitoring area. The total amount of 137Cs discharged directly into the ocean from the FDNPP has been estimated to be 3.6 ± 0.7 × 1015 Bq (Tsumune et al. 2013; Aoyama et al. 2016). Therefore, at least 1.3% of the directly discharged 137Cs was deposited in the area shallower than 800 m depth. The overall 137Cs inventory in the sediment must be significantly higher than the calculated surface abundance values owing to the presence of 137Cs at greater depth in the sediment column (see Sect. 3.6.2) and inside the 30-km radius from the FDNPP.
An exponential function fitted to the data from September 2011 to February 2016 yielded the following equation:
where t is the number of days after the accident (after 11 March 2011). We calculated the environmental halving time, including the physical decay of 137Cs, in the surface sediment in the monitoring area to be 2.32 years. The calculated value should be regarded as an upper limit because the calculation assumed that persistent input of 137Cs to the sediments, such as possible transport from the area close to the FDNPP and/or by rivers from the land, was negligible during the period considered. Using a three-box model consisting of the surface and bottom water and the bottom sediment, Watabe et al. (2013) evaluated the halving time of 137Cs in the surface sediment using data obtained from 1983 to 2010, before the accident, at eight sampling stations located about 30 km or more from the FDNPP and from the Fukushima Dai-ni Nuclear Power Plant (11 km south of the FDNPP). The removal rate of 137Cs from the sediment at each station was calculated to range from 1.2 × 10−4/day to 5.8 × 10−3/day; the halving time of 137Cs in the surface sediment before the accident based on the geometric mean of the rates was 2.22 years. Even though this calculation by Watabe et al. (2013) is based on a data set obtained at only eight sites with quite large variation and their model includes removal rates of 137Cs from the sediments and its input rates to the sediments as well, their halving time estimated from the removal rates agrees reasonably with ours.
By applying the pre-accident average concentration to the entire area of this study and the average water content and bulk density to each polygon, we estimated the abundance of 137Cs in the surface sediment before the accident as follows:
-
>30 km from the FDNPP: 6.9 × 1011 Bq.
-
<30 km: 0.5 × 1011 Bq.
-
total: 7.4 × 1011 Bq.
Admittedly, this pre-accident surface inventory is based on an oversimplified calculation, but it is reasonable to infer that the accident increased the surface 137Cs abundance in the sediments by two orders of magnitude.
3.6.2 Whole-core 137Cs inventory
We next calculated the 137Cs inventories and their temporal changes in the nine vertical profiles (Figs. 7, 11a). As can be predicted from the concentration variations, the inventories varied spatially by two orders of magnitude. In general, they decreased with time. While the surface concentration of 137Cs at Stn. I1 was the highest among all stations in February 2016 (see Figs. 4, 5), its whole-core inventory was also the highest among the cores studied.

a Temporal changes in the 137Cs inventory in the sediment column at nine sites. b Temporal changes in the surface-to-whole-inventory ratio (F 0–3) of 137Cs at the nine sites
The temporal trends of the whole-core inventory did not necessarily resemble the surface abundance trends. For example, among the nine stations, the highest rate of decrease in surface abundance, by a factor of 10, was at Stn. B3 from September 2012 to February 2016 (Fig. 8), whereas the whole-core inventory at that station changed by only a factor of two. This difference is probably attributable to the preferential removal of 137Cs from the surface, where it was more abundant than it was at depth.
3.6.3 Overall 137Cs inventory in the monitoring area
The presence of 137Cs in the sediment vertical profiles below the surface 3 cm, as shown by this (Fig. 7) and other studies (e.g., Ambe et al. 2015), suggests that the total sedimentary inventory in the monitoring area must be higher than the surface abundance. Because data on whole-core inventories are available from only nine stations, to estimate the whole-core inventory at other stations, we used the fraction of the whole-core inventory contained in the surface 3 cm of sediment, F 0–3 (Otosaka and Kato 2014). We therefore examined the temporal changes in the whole-core inventory and in the F 0–3 values at the nine stations (Fig. 11). As mentioned in Sect. 3.6.2, the whole-core inventory decreased with time to a variable extent at the nine stations (Fig. 11a). F 0–3 values also showed wide variability among stations, ranging from <0.1 to >0.8 (Fig. 11b). In general, at stations where the water depth was greater, 137Cs was more concentrated in the surface sediments than it was at stations where the water depth was shallower; in other words, 137Cs penetrated more deeply into the sediments at shallower water depths, probably owing to agitation of the surface sediment by vigorous movement of the overlying water by waves and tidal currents. We also observed that temporal changes in the F 0–3 values showed different trends among the stations (Fig. 11b), but the cause of those differences is not clear. The data for the inventory calculation are based on samples obtained by 2–3 sampling casts, each consisting of 16–24 sampling tubes; thus, these data should be relatively less sensitive to local spatial changes such as those shown by Thornton et al. (2013b). The temporal changes of F 0–3 may reflect dynamic features of the bottom sediment, such as deposition and removal of contaminated surface sediments. Future study is needed to clarify the reasons for the observed temporal changes of F 0–3.
To estimate the total 137Cs inventory in the sediments of the entire monitoring area, it is necessary to modify Eq. (1) to take account of the F 0–3 value, although, as Otosaka and Kato (2014) have pointed out, this seems to be impossible because the F 0–3 value is determined by multiple mechanisms. Overall, although F 0–3 varied over a wide range (Fig. 11b), it varied over a relatively smaller range from 0.2 to 0.4 in sediments from shallower water depths (0–200 m). Because most 137Cs derived from the FDNPP accident was in sediments at water depths of 0–200 m, we used 0.3 as a representative F 0–3 value. Thus, by dividing Eq. (1) by F 0–3 (i.e., 0.3), we reformulated the calculation of the 137Cs inventory in the monitoring area (excluding the area within a radius of 30 km from the accident site) as follows:
It is not easy to estimate the inventory of 137Cs within a 30-km radius from the power plant. TEPCO has been collecting bottom sediments and measuring radionuclides since right after the accident, but their method of retrieving sediments is different from that used in this study. According to Inatomi and Kusakabe (unpublished), who have attempted to use TEPCO’s data to estimate the surface abundance of 137Cs inside a 30-km radius from the FDNPP, the temporal trend of surface abundance within that radius seems to be not as linear as the trend at sites outside of the 30-km radius (see Fig. 10). Instead, the trend inside the 30-km radius seems to show three phases: during the initial phase, the 137Cs inventory decreased rapidly; this phase was followed by a phase during which the removal slowed; then, during the third phase, 137Cs increased. By using Eq. 2 and Inatomi and Kusakabe’s estimate, we estimated that the inventory in the entire monitoring area, including the area inside the 30-km radius, decreased from 230 × 1012 Bq in September 2011 to 54 × 1012 Bq in February 2016 (see Table 3 for details).
The calculated inventory for September 2011 accounted for about 6.4% of the direct release (3.6 × 1015 Bq; Tsumune et al. 2013; Aoyama et al. 2016). Another pathway by which 137Cs can reach the monitoring area is atmospheric input. The inputs to the near-FDNPP oceanic area, including both direct release and atmospheric input, have been estimated to be 11–27 × 1015 Bq, though the uncertainty is quite large owing to sparseness of the data set (Bailly du Bois et al. 2012; Charette et al. 2013; Rypina et al. 2013). Therefore, the percentage of 137Cs in the sediments relative to the total amount of 137Cs transported to the monitoring area in September 2011 is probably substantially lower than 6.4%. MacKenzie et al. (1994) suggested that about 10% of the 137Cs released from the Sellafield nuclear fuel reprocessing plant is taken up by Irish Sea sediment. If it is true that the Fukushima sediments contain a much lower fraction of sedimentary 137Cs than the Irish Sea sediments, the reason may be the differences in the sedimentary environment and the temporal pattern of 137Cs input into the Irish Sea. In particular, the interaction of the Sellafield discharges into the Irish Sea with fine-grained seafloor sediment (the Sellafield “mud patch”) can explain the 10% retention (MacKenzie et al. 1998). In addition, 137Cs was first released from the Sellafield plant in the 1960s, and the release has continued to the present, with variable annual quantities. The Cs concentration in the polluted water off Fukushima dropped by three orders of magnitude in a half year, a decrease that is attributable to the rapid exchange of coastal water with relatively uncontaminated open-ocean water. In contrast, Cs remains in solution in the Irish Sea for up to a year. Thus, the persistently high concentration of 137Cs in the Irish Sea water also leads to higher uptake by sediment there than off Fukushima.
Several attempts have been made, by different methods and for various areas and time periods, to estimate the 137Cs inventory off Fukushima (Table 3). Ambe et al. (2014) estimated the 137Cs inventory in the sediments to be 7.06 × 1013 Bq as of July 2012 outside a 20-km radius from the FDNPP and in the area bounded by 36°20′N, 37°50′N, 141°30′W, and the 500-m depth contour. Otosaka and Kato (2014) estimated the 134Cs inventory in the sediment by using data obtained during October–November 2011 and in October 2012. Here, we used their estimate to recalculate the 137Cs inventory to be 2.0 × 1014 Bq as of October 2011 in the area bounded by 35°40′N, 38°30′N, and the 800-m depth contour (25.5 × 103 km2), which approximately corresponds to the sampling area in this study (Table 3). Using several data sources for a wider area than that of this study, Black and Buesseler (2014) calculated 137Cs inventories in four zones off Fukushima for the period from June 2011 to September 2013. For this comparison, we selected three zones that when combined have an area of 24.1 × 103 km2, which is close to the area of this study, and recalculated the inventory in those zones to be ~1.2 × 1014 Bq. If the assumptions behind these calculations are taken into account, along with the fact that the data used were derived from different data sources obtained sometimes as much as a year apart, during which a significant temporal change would have occurred, the estimated inventories agree reasonably with our results. In contrast, Higashi et al. (2015) performed a numerical simulation by which they estimated the sedimentary 137Cs inventory to be 3.2 × 1015 Bq. Even though the area covered by the simulation (1.4 × 105 km2) is much greater than that of this study (2.7 × 104 km2), their estimated inventory is too high, almost equivalent to the total amount of 137Cs discharged directly from the FDNPP. Further study is needed to determine the reason for this high estimate.
3.7 Redissolution of sedimentary 137Cs from the sediments
As we indicated in Sect. 3.5, resuspension and subsequent lateral transport may play the most important role in reducing the 137Cs inventory in the sediment. Even so, the possibility that 137Cs is redissolved from the sediment into the water column cannot be ruled out. The redissolution of 137Cs from contaminated sediment has been reported, especially in the Irish Sea. For example, MacKenzie et al. (1998) have suggested that up to 77% of the 137Cs originally contained in the surface (<10 cm depth) sediment, and about 15% of that in deeper layers, was lost by redissolution between the late 1970s and 1992. Cook et al. (1997), by a mass balance calculation in that area, have estimated the halving time of the loss of Sellafield-derived 137Cs from the sediment by dissolution to be 23 years. Cesium is well known to have a strong affinity to clay (e.g., Nyffeler et al. 1984; Turner et al. 1993; Børretzen and Salbu 2002), and the behavior of clay-bound Cs should be the same as that of the clay particles. Possible pathways for the release of sedimentary Cs include desorption and ion exchange. Otosaka and Kobayashi (2013) performed a sequential extraction experiment with sediment collected off Ibaraki Prefecture, south of Fukushima Prefecture, and evaluated the contributions of the surface-adsorbed and organic matter-bound 137Cs to be less than 5% and about 20%, respectively, of total 137Cs. Their result indicates that organic material can be a major reservoir of labile 137Cs. In addition, the results of a dissolution experiment in which contaminated sediment was placed in uncontaminated seawater showed that more than 85% of the 137Cs remained in the sediments after 30 days (Otosaka and Kobayashi 2013). Ono et al. (2015) estimated that the ratio of the organic-bound 137Cs inventory to the total 137Cs inventory in the sediment off Fukushima ranges from 2.4 to 13.9%, with an average ratio (± standard deviation) of 5.7 (±2.9) %. Because we found a good correlation between ignition loss (≈ organic material) and silt and clay contents (the main medium of redistribution of sedimentary 137Cs) (Table 2), a part of the organic material was transported as particles. Nevertheless, 137Cs associated with organic material could potentially be desorbed and dissolved from the sediments.
A mass balance calculation of 137Cs in the water off Fukushima has shown that the 137Cs concentration in seawater outside of a 30-km radius from the FDNPP jumped from a pre-accident value of ~1.6 mBq/l to ~200 Bq/l in approximately 1 month and then decreased by three orders of magnitude (i.e., to ~0.1 Bq/l) by 6 months after the accident (Oikawa et al. 2013). However, the rate of decrease of the concentration subsequently fell, probably because of the continued release of 137Cs from the FDNPP, with the result that the concentration was still higher than the pre-accident level even 4 years after the accident. Kanda (2013) calculated that the continued 137Cs release averaged 9.3 × 1010 Bq/day in summer 2011 and 8.1 × 109 Bq/day in summer 2012. By a different approach, Tsumune et al. (2013) calculated similar post-accident release rates. If these estimates were affected by the dissolution of 137Cs from the sediment, how much of the 137Cs in seawater would have been derived from sediment? Takata et al. (2016), by using the fraction (15%) of 137Cs released from sediment obtained experimentally by Otosaka and Kobayashi (2013), estimated that the rate of 137Cs release from sediments decreased from 0.38 × 1012 to 0.14 × 1012 Bq/6 months in the 4 years after the accident in the area where the water depth was less than 300 m (~12,000 km2). Thus, the cumulative amount of 137Cs released from the sediment during those 4 years was estimated to be ~3 × 1012 Bq. If only the removal of 137Cs from surface sediment in 4.5 years (~3.5 × 1013 Bq; see Sect. 3.6.1) is considered, the estimated dissolution amount is one order of magnitude lower than the total removal amount. Takata et al. (2016) regarded 15% as the upper limit for the fraction of released 137Cs, because Otosaka and Kobayashi (2013) performed their experiment with relatively fresh sediments, which tend to release more 137Cs than sediment aged for, say, a few years or more. Therefore, the amount of 137Cs dissolved from the sediment may have been negligible in the monitoring area, at least during the period of monitoring (~5 years so far).
These results seem to be contradictory to the results of studies done in the Irish Sea (e.g., MacKenzie et al. 1998; Cook et al. 1997), which indicated that dissolution of 137Cs from the sediment is possible. Even if dissolution of 137Cs occurred in our monitoring area with a halving time of 23 years, the value calculated by Cook et al. (1997), it would not be detectable in our monitoring results owing to the overwhelmingly high removal by resuspension. The difference between the two regions may be attributable to the differences in the input mode and the oceanographic setting. Although the Sellafield plant has been releasing 137Cs and other radionuclides since the 1950s at a variable rate, most radionuclides from the FDNPP were released in a short period (~1 month). Further, the Irish Sea is a semi-enclosed sea, whereas the FDNPP faces the open ocean.
4 Concluding remarks
Five years of monitoring (May 2011–February 2016) of radioactivity in marine sediments revealed the spatiotemporal variation of 137Cs concentrations in the surface sediments. By almost a year after the accident, the 137Cs concentration in the surface sediments varied spatially by two orders of magnitude, probably reflecting variability in the sources of 137Cs to the area, such as the movement patterns of contaminated water and atmospheric plumes. Since then, the concentration has been decreasing with time to a variable extent. Accordingly, the 137Cs inventory in the sediment at each station has decreased as well. The overall inventory in the monitoring area, excluding the area within a 30-km radius from the FDNPP, decreased from September 2011 to February 2016 with a halving time of 2.3 years. Resuspension of bottom sediment and subsequent lateral transport appears to be the primary candidate mechanism of temporal change of 137Cs in the sediment. In other words, small, relatively mobile sedimentary particles such as silt, clay, and organic particles have been removed from the surface sediment and transported to areas with less bottom shear, where they have accumulated. Some 137Cs in the monitoring area has been resuspended and transported to outside the area, and some has been re-deposited within the area, with the result that both the spatial distribution of 137Cs and temporal changes in 137Cs are heterogeneous.
As a mechanism of 137Cs removal from the sediments, the possibility of 137Cs dissolution from the sediments into the water column cannot be excluded. However, a mass balance calculation (Takata et al. 2016) and our inventory calculation results suggest that dissolution of 137Cs from the sediment may not have contributed significantly to the 137Cs budget in the sediment. Although studies in the Irish Sea have shown redissolution of 137Cs from sediments to be an important mechanism (e.g., MacKenzie et al. 1998; Cook et al. 1997), different 137Cs behaviors in the two regions are attributable to differences in the input mode and in the oceanographic setting between them.
The results reported here are based mainly on data obtained from outside a 30-km radius from the FDNPP. Within that radius, the 137Cs concentration is assumed to be higher. Although we presented rough estimates for the 137Cs budget in sediment of the entire area around the FDNPP, including within the 30-km radius, evaluation of the overall inventory of FDNPP-derived 137Cs in sediment and its temporal change, utilizing data collected close to the accident site and taking account of subsequent secondary input to the area such as from rivers, is left for future work.
References
Adachi K, Kajino M, Zaizen Y et al (2013) Emission of spherical cesium-bearing particles from an early stage of the Fukushima nuclear accident. Sci Rep 3:2554. doi:10.1038/srep02554
Ambe D, Kaeriyama H, Shigenobu Y et al (2014) Five-minute resolved spatial distribution of radiocesium in sea sediment derived from the Fukushima Dai-ichi Nuclear Power Plant. J Environ Radioact 138:264–275
Ambe D, Kaeriyama H, Shigenobu Y et al (2015) Three-dimensional distribution of radiocesium in sea sediment derived from the Fukushima Dai-ichi Nuclear Power Plant. In: Nakata K, Sugisaki H (eds) Impacts of the Fukushima nuclear accident on fish and fishing grounds. Springer Japan, Tokyo, pp. 53–65. doi:10.1007/978-4-431-55537-7_4
Aoyama M, Tsumune D, Uematsu M et al (2012) Temporal variation of 134Cs and 137Cs activities in surface water at stations along the coastline near the Fukushima Dai-ichi Nuclear Power Plant accident site, Japan. Geochem J 46:321–325
Aoyama M, Kajino M, Tanaka TY et al (2016) 134Cs and 137Cs in the North Pacific Ocean derived from the March 2011 TEPCO Fukushima Dai-ichi Nuclear Power Plant accident, Japan. Part two: estimation of 134Cs and 137Cs inventories in the North Pacific Ocean. J Oceanogr 72:67–76. doi:10.1007/s10872-015-0332-2
Bailly du Bois P, Laguionie P, Boust D et al (2012) Estimation of marine source-term following Fukushima Dai-ichi accident. J Environ Radioact 114:2–9
Black EE, Buesseler KO (2014) Spatial variability and the fate of cesium in coastal sediments near Fukushima, Japan. Biogeosciences 11:5123–5137
Børretzen P, Salbu B (2002) Fixation of Cs to marine sediments estimated by a stochastic modeling approach. J Environ Radioact 61:1–20
Buesseler KO, Jayne SR, Fisher NS et al (2012) Fukushima-derived radionuclides in the ocean and biota off Japan. PNAS 109(16):5984–5988
Buesseler KO, German CR, Honda MC et al (2015) Tracking the fate of particle associated Fukushima Daiichi Cesium in the Ocean off Japan. Environ Sci Technol 49:9807–9816
Charette MA, Breier CF, Henderson PB et al (2013) Radium-based estimates of cesium isotope transport and total direct ocean discharges from the Fukushima Nuclear Power Plant accident. Biogeosciences 10:2159–2167
Cook GT, MacKenzie AB, McDonald P et al (1997) Remobilization of sellafield-derived radionuclides and transport from the North-east Irish Sea. J Environ Radioact 35(3):227–241
Higashi H, Morino Y, Furuichi N et al (2015) Ocean dynamic processes causing spatially heterogeneous distribution of sedimentary caesium-137 massively released from the Fukushima Daiichi Nuclear Power Plant. Biogeosciences 12:7107–7128
Huang S-W (1982) Study on coastal trawl in Senday Bay and its adjacent waters. II. Fishing effort and fishing power. Tohoku J Agr Res 33:22–41
Ikehara K, Irino T, Usami K et al (2014) Possible submarine tsunami deposits on the outer shelf of Sendai Bay, Japan resulting from the 2011 earthquake and tsunami off the Pacific coast of Tohoku. Mar Geol 358:120–127
Ikenoue T, Takata H, Kusakabe M et al (2017) Temporal variation of cesium isotope concentrations and atom ratios in zooplankton in the Pacific off the east coast of Japan. Sci Rep 7:39874. doi:10.1038/srep39874
Japanese Industrial Standard (2009) Test method for particle size distribution of soils. JIS A 1204
Kanda J (2013) Continuing 137Cs release to the sea from the Fukushima Dai-ichi Nuclear Power Plant through 2012. Biogeosciences 10:6107–6113
Kusakabe M, Oikawa S, Takata H et al (2013) Spatiotemporal distributions of Fukushima-derived radionuclides in nearby marine surface sediments. Biogeosciences 10:5019–5030
MacKenzie AB, Scott RD, Allan RL et al (1994) Sediment radionuclide profile: implications for mechanisms of Sellafield waste dispersal in the Irish Sea. J Environ Radioact 23(1):39–69
MacKenzie AB, Cook GT, McDonald P et al (1998) The influence of mixing timescales and re-dissolution processes on the distribution of radionuclides in the northeast Irish Sea sediments. J Environ Radioact 39(1):35–53
Misumi K, Tsumune D, Tsubono T et al (2014) Factors controlling the spatiotemporal variation of 137Cs in seabed sediment off the Fukushima coast: implications from numerical simulations. J Environ Radioact 136:218–228
Noguchi T, Tanikawa W, Hirose T et al (2012) Dynamic process of turbidity generation triggered by the 2011 Tohoku-Oki earthquake. Geochem Geophys Geosyst. doi:10.1029/2012GC004360
Nyffeler UP, Li YH, Santschi PH (1984) A kinetic approach to describe trace-element distribution between particles and solution in natural aquatic systems. Geochim Cosmochim Acta 48:1513–1522
Oikawa S, Takata H, Watabe T et al (2013) Distribution of the Fukushima-derived radionuclides in seawater in the Pacific off the coast of Miyagi, Fukushima, and Ibaraki Prefectures, Japan. Biogeosciences 10:5031–5047
Ono T, Ambe D, Kaeriyama H et al (2015) Concentration of 134Cs + 137Cs bonded to the organic fraction of sediments offshore Fukushima, Japan. Geochem J 49:219–227
Otosaka S, Kato Y (2014) Radiocesium derived from the Fukushima Daiichi Nuclear Power Plant accident in seabed sediments: Initial deposition and inventories. Environ Sci Process Impact 6(5):978–990. doi:10.1039/C4EM00016A
Otosaka S, Kobayashi T (2013) Sedimentation and remobilization of radiocesium in the coastal area of Ibaraki, 70 km south of the Fukushima Dai-ichi Nuclear Power Plant. Environ Monit Assess 185(7):5419–5433. doi:10.1007/s10661-012-2956-7
Rypina II, Jayne SR, Yoshida S et al (2013) Short-term dispersal of Fukushima-derived radionuclides off Japan: modeling efforts and model-data intercomparison. Biogeosciences 10:4973–4990
Seike K, Kitahashi T, Noguchi T (2016) Sedimentary features of Onagawa Bay, northeastern Japan after the 2011 off the Pacific coast of Tohoku Earthquake: sediment mixing by recolonized benthic animals decreases the preservation potential of tsunami deposits. J Oceanogr 72(1):141–149
Takata H, Hasegawa K, Oikawa S et al (2015) Remobilization of radiocesium on riverine particles in seawater: the contribution of desorption to the export flux to the marine environment. Mar Chem 176:51–63
Takata H, Kusakabe M, Inatomi N et al (2016) The contribution of sources to the sustained elevated inventory of 137Cs in offshore waters east of Japan after the Fukushima Dai-ichi Nuclear Power Station accident. Environ Sci Technol 50:6957–6963
Thornton B, Ohnishi S, Ura T et al (2013a) Continuous measurement of radionuclide distribution off Fukushima using a towed sea-bed gamma ray spectrometer. Deep Sea Res Part I 79:10–19
Thornton B, Ohnishi S, Ura T et al (2013b) Distribution of local 137Cs anomalies on the seafloor near the Fukushima Dai-ichi Nuclear Power Plant. Mar Pollut Bull 74:344–350
Tsumune D, Tsubono T, Aoyama M et al (2013) One-year, regional-scale simulation of 137Cs radioactivity in the ocean following the Fukushima Dai-ichi Nuclear Power Plant accident. Biogeosciences 10:5601–5617
Turner A, Millward GE, Bale AJ et al (1993) Application of the K D concept to the study of trace metal removal and desorption during estuarine mixing. Estuar Coast Shelf S 36:1–13
Watabe T, Oikawa S, Isoyama N et al (2013) Spatiotemporal distribution of 137Cs in the sea surrounding Japanese Islands in the decades before the disaster at the Fukushima Daiichi Nuclear Power Plant in 2011. Sci Total Environ 463–464:913–921
Yagi H, Sugimatsu K, Kawamata S et al (2015) Bottom turbidity, boundary layer dynamics, and associated transport of suspended particulate materials off the Fukushima Coast. In: Nakata K, Sugisaki H (eds) Impacts of the Fukushima nuclear accident on fish and fishing grounds. Springer Japan, Tokyo, pp. 77–89. doi:10.1007/978-4-431-55537-7_6
Acknowledgements
The data presented here were obtained under contract with the Ministry of Education, Culture, Sports, Science and Technology from March 2011 to March 2013 and with the Nuclear Regulation Authority from April 2013 to March 2014. We thank the staff members of the Marine Ecology Research Institute for their great help with the program logistics and fieldwork.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kusakabe, M., Inatomi, N., Takata, H. et al. Decline in radiocesium in seafloor sediments off Fukushima and nearby prefectures. J Oceanogr 73, 529–545 (2017). https://doi.org/10.1007/s10872-017-0440-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10872-017-0440-2