
Abstract
A new polymorph of tert-butyl (2-aminophenyl)carbamate was identified using single crystal X-ray diffraction. The compound crystallised in the centrosymmetric, monoclinic space group P21/n with an asymmetric unit comprising two crystallographically-independent molecules (Zʹ = 2). This new structure was compared to that of the known polymorph with the differences between the two being attributed to a combination of space group symmetry, conformational variation, hydrogen bonding network dimensionality and crystal packing.
Index Abstract
A new polymorph of tert-butyl (2-aminophenyl)carbamate exhibits π···H interactions that precipitate conformational variations and an asymmetric unit with two crystallographically-independent molecules.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ortho-Phenylenediamine is a versatile reagent finding many uses in synthetic chemistry e.g. as a redox-active ligand [1], a monomer building block to create conducting polymers [2] or as a precursor to the synthesis of benzimidazole heterocyclic compounds possessing broad spectrum disease activity [3]. Our interest in this molecule stems from the latter, more specifically the mono-protected analogue, tert-butyl (2-aminophenyl)carbamate), and its use in the synthesis of novel benzimidazoles that could potentially replace the benzoxa- [1,2,3]-diazoles in some potent anti-tubercular compounds [4].

tert-butyl (2-aminophenyl)carbamate with the numbering scheme used in this article
The crystal structure of tert-butyl (2-aminophenyl)carbamate was reported in 2014 by Singh and co-workers (CSD Refcode: TUGMIT) [5]. The structure crystallised in the non-centrosymmetric, monoclinic P21 space group with an asymmetric unit comprising one molecule (Zʹ = 1). For the purposes of this study, this structure will be referred to as polymorph I.
In this work we report the discovery of a new polymorph of tert-butyl (2-aminophenyl)carbamate (polymorph II) and compare the structure with that of the known polymorph (polymorph I) in terms of conformation, intermolecular interactions and packing similarity.
Experimental
tert-Butyl (2-aminophenyl)carbamate was prepared according to literature methods [6]. Crystals of polymorph II suitable for single crystal X-ray diffraction were grown as colourless plates by slow evaporation of the solvent from a solution of the compound in dichloromethane. The melting point range for crystals of polymorph II was determined to be 386–388 K.
Single crystal diffraction on an XtaLAB Synergy HyPix-Arc 100 diffractometer using copper radiation (λCuKα = 1.54184 Å). Data were collected at 150 K using an Oxford Cryosystems CryostreamPlus open-flow N2 cooling device.
Intensities were corrected for absorption using a multifaceted crystal model created by indexing the faces of the crystal for which data were collected [7]. Cell refinement, data collection and data reduction were undertaken via the software CrysAlisPro [8].
All structures were solved using XT [9] and refined by XL [10] using the Olex2 interface [11]. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were positioned with idealised geometry, with the exception of those bound to heteroatoms, the positions of which were located using peaks in the Fourier difference map. The displacement parameters of the hydrogen atoms were constrained using a riding model with U(H) set to be an appropriate multiple (1.2 or 1.5) of the Ueq value of the parent atom.
Results and Discussion
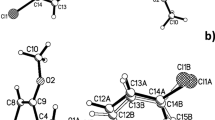
The structure of tert-butyl (2-aminophenyl)carbamate is the second polymorph (polymorph II) of a known structure (polymorph I), for which data are available in the Cambridge Structural Database (CSD Refcode: TUGMIT) [5]. Where polymorph I crystallises in the non-centrosymmetric monoclinic space group P21 with one molecule in the asymmetric unit (Z′ = 1), polymorph II crystallises in the centrosymmetric monoclinic space group P21/n with an asymmetric unit comprising two molecules (Z′ = 2) (Fig. 2). Details of the refinements for both structures are presented in Table 1.

The asymmetric unit of polymorph II with ellipsoids drawn at the 50% probability level. Hydrogen atoms not involved in hydrogen bonding have been omitted for clarity
A comparison of the bond distances of the two molecules reveals that there are no significant deviations between the two. The presence of two crystallographically-independent molecules in the asymmetric unit of polymorph II can be attributed to a slight conformational variation between the two molecules. One of the molecules of polymorph II (molecule 2) is almost identical in terms of conformation to that of polymorph I with an RMSD of 0.0419 Å calculated for an overlay of the two. By way of contrast, the other molecule in the asymmetric unit of polymorph II exhibits noticeably different conformation, with a RMSD of 0.2183 Å measured when overlayed with polymorph I (Fig. 3).

Overlay diagram of polymorph I (red) and molecule 2 of polymorph II (yellow)
These different conformations can in turn be attributed to variations in the torsion angles about the carbonyl bond in each of the molecules (Table 2). As is reflected in the low RMSD measured for the overlay between polymorph I and molecule 2 of polymorph II, the torsion angles beginning with the carbonyl bond are within error for these molecules. The same cannot be said for molecule 1 of polymorph II for which the angles are observed to differ significantly.
The difference in the asymmetric units and space group symmetry of the two polymorphs also has implications for their packing. This can be demonstrated through analysis of the hydrogen bonding networks in both structures. In the structure of polymorph I the hydrogen bonding network is a one-dimensional motif formed of bifurcated N-H···O interactions involving two donors and one acceptor. The overall effect is a chain of 7-membered ring hydrogen bonding motifs propagating in the crystallographic [100] direction with each adjacent molecule related by translation symmetry (Fig. 4). Using Margaret Etter’s graph set notation for hydrogen bonding networks, this chain of rings can be characterised as a C(4)C(7)[R21(7)] motif [12]. It is worth noting that one of the protons of the primary amine group is not involved in hydrogen bonding.

The one-dimensional hydrogen bonding motif along the [100] direction in the structure of polymorph I viewed down the [0–11] direction with hydrogen bonds depicted as dashed lines. Hydrogen atoms not involved in hydrogen bonding have been omitted for clarity
The hydrogen bonding network in the structure of polymorph II is more complex than that of polymorph I (Table 3). The motif in this case is an alternating hydrogen bonded chain of molecule 1 and molecule 2. The bifurcated hydrogen bond forming a 7-membered ring is observed in both structures, however, the chain motif incorporating this ring is different for polymorph II. Where in the structure of polymorph I there is a 7-membered hydrogen-bonded ring linking each molecule in the chain, it is only observed between every second molecule in the equivalent chain in polymorph II (Fig. 5).

The one-dimensional hydrogen bonding motif along the [010] direction in the structure of polymorph II with molecular numbering scheme as viewed approximately down the [100] direction. Hydrogen bonds and the N-H···π interactions are depicted as dashed lines. The unit cell axes and hydrogen atoms not involved in hydrogen bonding have been omitted for clarity
As adjacent molecules in this chain are not related by crystallographic symmetry, they are at slightly different orientations relative to one another and this precludes the formation of the 7-membered ring between every molecule; the ring motif forms only where molecule 1 donates to molecule 2. This hydrogen bonded chain propagating in the [010] direction with the symmetry of the 21 screw axis, can be described as a C22(8)C22(11)[R21(7)] motif using graph set notation.
Interestingly, in cases where the primary amine group of molecule 2 is not involved in the bifurcated interaction, a close N-H···π interaction is observed between this amine group and the benzene ring of molecule 1 (Fig. 5). This interaction appears to be the origin of the two different molecular conformations in the structure of polymorph II, as this attractive electrostatic interaction pulls the benzene ring of molecule 1 towards the primary amine group of molecule 2. This perturbation results in the change in torsion angle observed in molecule 1, which is the salient difference between the conformations of the two molecules in polymorph II (Table 2).
In addition to the chains of rings in the [010], further hydrogen bonding is observed in the [100] direction in the structure of polymorph II. In this direction, the molecules are linked by two hydrogen bonds accepted by the carbonyl oxygen atom of molecule 1 from a proton of the primary amine group of molecule 2 and by the secondary amine group of an adjacent identical molecule. These interactions form a further chain of hydrogen bonds in the [100] direction with the graph set C21(7). As the chains in the [010] and [100] directions are linked together this results in a two-dimensional hydrogen bonded network co-planar to the crystallographic (001) plane (Fig. 6).

The two-dimensional hydrogen bonding network co-planar with the (001) plane in the structure of polymorph II with molecular numbering as viewed down the [001] direction. Hydrogen bonds are depicted as dashed lines. The N-H···π interactions and hydrogen atoms bound to carbon atoms have been omitted for clarity
Although it is clear from an analysis of the unit cell parameters, space group symmetry, molecular conformations and hydrogen bonding networks of the two structures that they are unequivocally two different polymorphs, a Crystal Packing Similarity analysis can provide further demonstration of this. This analysis can be performed using the Crystal Packing Similarity feature in the software Mercury developed by the CCDC [13]. To allow for the comparison, a cluster of 15 molecules was selected for each structure and using the default geometric tolerances (20% for distances and 20° for the angles) the two structures were combined to maximise the number of molecules that overlay.
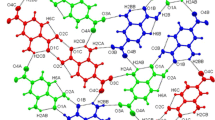
The results of this comparison show that only 1 of the 15 molecules in each cluster were able to overlap. A visual analysis of how different the packing is around the one shared molecule gives a stark demonstration of just how drastically different the two structures are (Fig. 7).

The packing similarity of polymorph I (yellow) and polymorph II (red). The two molecules found to overlap are rendered in blue
Conclusion
The crystal structure of polymorph II of tert-butyl (2-aminophenyl)carbamate has been reported and compared to the known form, polymorph I. A comparison of the two structures revealed the ways in which the two structures differ from each other. Polymorph I crystallises in the non-centrosymmetric P21 space group with one molecule in the asymmetric unit (Zʹ = 1) where polymorph II crystallises in the centrosymmetric P21/n and a Zʹ value of 2. One of the molecules in the asymmetric unit was found to adopt a different conformation to the molecules in polymorph I.
The two structures exhibited different hydrogen bonding motifs with a simple chain of rings being observed in polymorph I where a more complex two-dimensional network forms in the structure of polymorph II. The analysis of the hydrogen bonding also revealed that the origin of the conformational variation is a molecular perturbation resulting from N-H···π interactions. A Crystal Packing Similarity analysis further demonstrated the differences in the supramolecular structures of both polymorphs.
Data Availability
CCDC 2352341 contains the full crystallographic data for this article. These are available free of charge at www.ccdc.cam.ac.uk/data_request/cif, by emailing data_request@ccdc.cam.ac.uk or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK; fax: +44 1223 336033.
References
Broere DLJ, Plessiusa R, Vlugt JI (2015) New avenues for ligand-mediated processes – expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem Soc Rev 44:6886
Ergun C (2023) A current review on conducting PolymerBased catalysts: advanced oxidation processes for the removal of aquatic pollutants. Water Air Soil Pollut 234:524
Mohammed LA, Farhan MA, Dadoosh SA, Alheety MA, Majeed AH, Ali Saadon Mahmood AS, Mahmouda ZH (2023) Review on Benzimidazole Heterocyclic compounds: synthesis and their Medicinal Activity Applications. SynOpen 7:652
Brown AK, Aljohani AKB, Gill JH, Steel PG, Sellars JD (2019) Identification of Novel Benzoxa-[2,1,3]-diazole substituted amino acid hydrazides as potential Anti-tubercular agents. Molecules 24:811
Singh MK, Gangwar M, Kumar D, Tilak R, Nath G, Agarwal A (2014) In vitro antimicrobial activity of o-phenylenediamine-tert-butyl-N1,2,3-triazole carbamate analog. Med Chem Res 23:4962
Stachura LM, Logsdon PB, Swan EL, Basu R (1992) US Patent 5,085,796
Clark RC, Reid JS (1995) The analytical calculation of absorption in multifaceted crystals. Acta Cryst A51:887
CrysAlisPro R Oxford Diffraction, Tokyo, Japan
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr Sect A: Found Crystallogr 71:3
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr Sect A: Found Crystallogr 64:112
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst 42:339
Etter MC (1990) Encoding and decoding hydrogen-bond patterns of Organic compounds. Acc Chem Res 23:121
Macrae CF, Sovago I, Cottrell SJ, Galek PTA, McCabe P, Pidcock E, Platings M, Shields GP, Stevens JS, Towler M, Wood PA (2020) Mercury 4.0: from visualization to analysis, design and prediction. J Appl Cryst 53:226
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [Grant number EP/S022791/1].
Author information
Authors and Affiliations
Contributions
Z.E.P. and M.T.H prepared the title compound and grew the crystals under the supervision of J.D.S. P.G.W. collected and refined the structure. P.G.W. and J.D.S. wrote the manuscript which was reviewed by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Potter, Z.E., Hill, M.T., Sellars, J.D. et al. A New Polymorph of tert-Butyl (2-Aminophenyl)Carbamate. J Chem Crystallogr (2024). https://doi.org/10.1007/s10870-024-01021-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10870-024-01021-6