
Abstract
In recent years, many solid-state hydride-based materials have been considered as hydrogen storage systems for mobile and stationary applications. Due to a gravimetric hydrogen capacity of 5.6 wt% and a dehydrogenation enthalpy of 38.9 kJ/mol H2, Mg(NH2)2 + 2LiH is considered a potential hydrogen storage material for solid-state storage systems to be coupled with PEM fuel cell devices. One of the main challenges is the reduction of dehydrogenation temperature since this system requires high dehydrogenation temperatures (~ 200 °C). The addition of KH to this system significantly decreases the dehydrogenation onset temperature to 130 °C. On the one hand, the addition of KH stabilizes the hydrogen storage capacity. On the other hand, the capacity is reduced by 50% (from 4.1 to 2%) after the first 25 cycles. In this work, the particle sizes of the overall hydride matrix and the potassium-containing species are investigated during hydrogen cycling. Relation between particle size evolution of the additive and hydrogen storage kinetics is described by using an advanced synchrotron-based technique: Anomalous small-angle X-ray scattering, which was applied for the first time at the potassium K-edge for amide-hydride hydrogen storage systems. The outcomes from this investigation show that, the nanometric potassium-containing phases might be located at the reaction interfaces, limiting the particle coarsening. Average diameters of potassium-containing nanoparticles double after 25 cycles (from 10 to 20 nm). Therefore, reaction kinetics at subsequent cycles degrade. The deterioration of the reaction kinetics can be minimized by selecting lower absorption temperatures, which mitigates the particle size growth, resulting in two times faster reaction kinetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen storage technologies can provide a solution to our environmental challenges, which are caused mainly by our heavy reliance on hydrocarbons[1]. Compared to gaseous H2 storage at 700 bar or liquid H2 storage at − 253 °C, amide-based solid-state hydrogen storage devices have the advantage of functioning at a moderate temperature (from r.t. to 200 °C) and pressure (below 100 bar). Chen et al. [2] developed the first reversible amide-hydride hydrogen storage system. Li3N was claimed to be able to store more than 10 wt% H2 based on the following reaction:
However, according to Eq. (1), the enthalpy of the reaction of LiNH2 with LiH to form Li2NH is 66 kJ/mol H2, which is still too high for mobile applications [3]. When Mg2+ replaces Li+ in LiNH2, Mg(NH2)2 + 2LiH system with better thermodynamic characteristics is formed. This system possesses a gravimetric hydrogen capacity of 5.6 wt% and—because of a dehydrogenation enthalpy of 38.9 kJ/mol H2—theoretically allows for hydrogen desorption near 90 °C at 1 bar H2 [4]. The dehydrogenation process occurs in two steps, as described in Eqs. (2) and (3) [5]:
Despite the Mg(NH2)2 + 2LiH system's fascinating thermodynamics, experimentally significant dehydrogenation rates can only be achieved at temperatures above 200 °C due to severe kinetic barriers [6]. As previously stated, the dehydrogenation reaction occurs in two steps. The poor reaction kinetics are caused by species diffusion at the amide-hydride contacts, as well as nucleation and development of imide phases [7]. In order to accelerate the reaction kinetics, several additives have been investigated so far [8,9,10,11,12,13,14,15]. K-based compounds have consistently been one of the most effective additives [16]. Wang et al. [8] reported that the addition of 3 mol% KH decreases the desorption temperature of Mg(NH2)2 + 2LiH by about 50 °C (from 180 to 130 °C) and enhances the kinetic behaviour. The interaction of KH with Mg(NH2)2 leads to the formation of intermediate phase K2Mg(NH2)4 with a lower nucleation barrier, this newly formed phase metathesizes rapidly with LiH to regenerate KH. [16]. Three K-containing species, i.e. KH, K2Mg(NH2)4, and Li3K(NH2)4 have been proposed to be responsible for the improved reaction kinetics [17].
The operating temperature drastically influences the reaction kinetics and structural properties of Mg(NH2)2 + 2LiH with K-based additives. Although dehydrogenation kinetics accelerate with increasing temperature at first, this results in unfavourable dehydrogenation/hydrogenation characteristics in the following cycles [18]. Two main explanations have been proposed for the poor performance at high temperatures; growth of particle and grain sizes of the K-species upon dehydrogenation and their inhomogeneous distribution. Moreover, above 180 °C the structural transformation of the dehydrogenation product Li2Mg(NH)2 from cubic to orthorhombic phase may also contribute to the sluggish kinetic behaviour, since the hydrogenation temperature of the cubic phase is lower than the orthorhombic phase [18].
The structural and kinetic properties of the Mg(NH2)2 + 2LiH + xKH (x: 0–0.3) system are investigated in this paper. Structural properties are studied with advanced synchrotron techniques such as in-situ powder X-ray diffraction (SR-PXD), and with a combination of small-angle X-ray scattering (SAXS) and anomalous small-angle X-ray scattering (ASAXS). The hydrogen kinetic properties are assessed by high-pressure differential scanning calorimetry (HP-DSC) and Sieverts apparatus. This investigation provides new insight on the correlation between particle sizes, reaction kinetics, and dehydrogenation/hydrogenation temperatures of the Mg(NH2)2 + 2LiH reactive hydride composite with KH addition.
Experimental
LiH (Alfa Aesar, 99.9%) and KH (suspension 35% in mineral oil) were purchased from Sigma-Aldrich. To remove the mineral oil, the KH-oil mixture was washed 3× with Pentane and dried inside a glove box. Mg(NH2)2 was synthesized in-house via a 2-step process. Firstly, MgH2 was ball milled at 400 rpm and under 7 bar of NH3 pressure for 24 h. The milling procedure was performed in a Fritsch P6 planetary mill device. Then, the as-milled material was heated to 300 °C under 7 bar of NH3 in a pressure reactor to increase the yield of Mg(NH2)2·Mg(NH2)2 with 95% purity was obtained.
Appropriate amounts of the starting materials were mixed and ball milled at 400 rpm for 36 h under 50 bar of H2. In total, five samples were prepared. To prevent contamination, all powders handling and milling were performed in an MBraun glove box filled with Ar with H2O and O2 levels below 10 ppm.
As-milled samples were subjected to heat and gas pressure treatment in an autoclave from Estanit GmbH. Six different sample compartments allowed reproducible absorption/desorption reactions for different compositions of materials. About 400 mg of each sample were placed in the individual compartments. The autoclave was first evacuated and then filled with 50 bar of H2. Then, it was heated up to 180 °C to obtain a final hydrogen pressure of about 80 bar. Under isothermal conditions, absorption/desorption reactions were performed by setting a hydrogen pressure of 1 bar for 20 h and 80 bar for 4 h. These conditions were repeated 25× to cycle all samples at once.
Differential scanning calorimetry (DSC) measurements were carried out using a Netsch DSC TASC 414/4 calorimeter which was kept in an argon-filled glovebox. Roughly 5 mg of sample were placed into an Al2O3 crucible and heated from room temperature (RT) to 300 °C with a heating rate of 3 °C/min. In order to have the same conditions as in the volumetric measurements, pressures for dehydrogenation and re-hydrogenation were selected as 1 bar of H2 and 80 bar of H2, respectively. Using magnetic valves, we were able to eliminate the effect of temperature on the set pressure with a tolerance of ± 0.2 bar. The DSC chamber was filled with Ar while placing the sample crucible. The chamber was flushed with hydrogen gas three times to release the remaining Ar. To calculate activation energies, different heating ramps were used: 1 °C/min, 3 °C/min, 5 °C/min and 10 °C/min.
Ex-situ powder X-ray diffraction (PXD) patterns were collected using a Bruker D8 Discover diffractometer with a Cu X-ray source (λ = 1.54184 Å) and a 2D detector in Bragg–Brentano geometry. The diffraction patterns were plotted as a function of the scattering vector q(nm−1) to have wavelength-independent data. Samples were protected from air and moisture during the measurement with the use of a PMMA dome sample holder produced by Bruker.
In-situ synchrotron radiation powder, X-ray diffraction (SR-PXD) experiments were performed at MAX II Synchrotron facility in Lund, Sweden, at the beamline I711 [19]. The instrumental geometry of the powder diffractometer is a Debye-Scherrer like configuration with a selected wavelength λ ~ 0.99 Å and a plate image Agilent Titan detector (2048 × 2048 pixel, each of size 60 × 60 mm2) placed about 80 mm from the sample holder. The calibration of the diffraction line positions was carried out using a LaB6 powder. The powder was introduced into a quartz capillary tube inside an Argon-filled glove box and then fixed on an in-house developed in-situ cell [20], which allows controlling the heating temperatures and operating pressures [21]. First, dehydrogenation was recorded from RT to 180 °C, with a heating rate of 5 °C min−1 under 1 bar of H2. Following this experiment, the first hydrogenation was recorded from RT to 180 °C under 80 bar of H2 with a heating ramp of 5 °C min−1. Then, the second dehydrogenation was recorded from r.t. to 180 °C. The SR-PXD data were reported referring to intensity against the scattering vector q = 4πsinθ/λ (nm−1), where λ is the X-ray wavelength and θ is the scattering angle. The 2D image was integrated by FIT2D software. Quantitative analyses were performed using MAUD software implementing the Rietveld method [22].
(Anomalous) Small-angle X-ray scattering ((A)SAXS) curves of the as-milled and cycled samples were collected using the HZB SAXS/ASAXS instrument at FCM-beamline of PTB, installed at synchrotron facility BESSY II (HZB, Germany) [23]. The four-crystal monochromator has an energy resolution of ΔE/E ~ 2 × 10–4, while using the Si111 crystals. An in-vacuum version of a 1 M Pilatus detector was used to record the data [24]. All measurements were carried out at 2 sample-to-detector distances (~ 0.8 m and ~ 3.8 m) to cover the maximum possible experimental q-range. Here, q is the magnitude of the scattering vector defined above. 10 keV were chosen as X-ray energy in the case of SAXS. Samples for SAXS/ASAXS measurements were prepared in N2-filled glove box at the EMIL Lab at BESSY. Each sample was packed between Kapton tapes from each side to prevent the contamination with air and moisture during the sample transfer to the SAXS beamline. The prepared samples were fixed at molybdenum sample holder sheets of 0.2 mm thickness with a circular hole of 5 mm in diameter. ASAXS measurements were carried out at six different energies close and below the potassium K-edge at 3610 eV (3608, 3603, 3587, 3537, 3453, and 3353 eV), in order to obtain scattering contrast. The scattering data were corrected for the scattering of the Kapton foils and normalized to absolute scattering cross sections as reported elsewhere [25, 26]. X-ray absorption near edge structure (XANES) spectroscopy was measured according to find the potassium absorption edge of each sample using the same set-up.
The volumetric differential pressure method was used to determine the hydrogen storage capacity of as-milled and cycled composites. An in-house made Sieverts apparatus was utilized for this purpose. About 100 mg of sample was loaded into the sample holder. All desorption and absorption measurements were performed under 1 bar of H2 and under 80 bar of H2, respectively, starting from RT to 180 °C with a heating rate of 3 °C min−1.
Results and discussion
Thermal behaviour and reaction kinetics
Thermal behaviours during the first dehydrogenation of the as-milled samples with and without KH were determined through DSC, as shown in Fig. 1. All samples exhibit endothermic dehydrogenation events up to 260 °C. Dehydrogenation of the \({\text{Mg}}{\left({\text{NH}}_{2}\right)}_{2}+ \text{2LiH}\) sample displays a peak around 190 °C with a shoulder at 200 °C, suggesting a 2-step reaction mechanism as stated in Eqs. 2 and 3. The peak positions of these two reactions shift to lower temperatures when KH is introduced. Small amount of KH addition (0.07 mol) is sufficient to lower the peak temperature of the 1st step of the reaction from 190 to 140 °C. Peak temperature of the second reaction step decreases gradually with an increasing amount of KH (from 200 to 155 °C by adding 0.15 mol of KH). In the case of sample containing 0.30 mol KH, these two events possibly occur at similar temperatures around 145 °C, reaction pathway is not altered.

DSC curves of as-milled samples
In order to evaluate the effect of additive on the material kinetic behaviour, activation energies from the 1st dehydrogenation reactions using the differential scanning calorimeter under 1 bar of H2 pressure with varying heating rates (1, 3, 5, and 10 °C/min) were determined with Kissinger’s method according to Eq. (4):
where Tm is the peak temperature at the maximum reaction rate, β the heating rate, ‘A’ the pre-exponential factor, R the gas constant, and Ea the apparent activation energy. The slope of the plot ln(β/Tm2) against 1/Tm provides the value of Ea/RT, and the intercept corresponds to ‘A’. The activation energies are determined from the peak maxima, where the reaction rate is maximum. Figure 2 shows the Kissinger plots of the as-milled samples during the 1st dehydrogenation reaction (see supplementary content, ESI Figs. 1–4 for original DSC curves).

Kissinger plots of as-milled samples measured at 1 bar of H2 pressure and different heating rates (1, 3, 5, and 10 °C/min)
All results obtained from the Kissinger plots are summarized in Table 1. The pristine sample has a dehydrogenation activation energy Ea of 168 ± 4 kJ/mol. Even though the dehydrogenation peak temperatures are lowered by KH addition (Fig. 1), Ea is higher for all samples containing KH as shown in Table 1. The calculated Ea increases by nearly 50 kJ/mol H2 for the sample containing 0.07 mol and reaches 215 ± 9 kJ/mol H2, if compared with the pristine sample. Interestingly, the pre-exponential factor A, for the samples containing potassium hydride is much larger than that for the pristine one. In order to understand these contradictory findings, rate constants (k) were calculated by the Arrhenius expression k = A∙exp[− Ea/RT] (1/s) at the cycling temperature of 180 °C (see Fig. 3). Calculated rate constants from the Arrhenius expression indicate that KH-containing samples have a larger rate constant compared to the pristine sample at 180 °C. A noticeable increase of the frequency factor for potassium-containing samples leads to a more effective interaction between the reactants. This interaction is slightly improved with increased KH amount, since the rate constant values are rising gradually. These outcomes agree with the results from our previous work, where another potassium compound is used as an additive: K2Mn(NH2)4 [11].

Reaction kinetics at a 1st dehydrogenation b 25th dehydrogenation
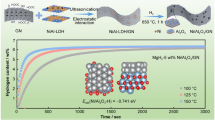
Dehydrogenation properties of the as-milled and cycled materials were determined by Sieverts apparatus, as shown in Fig. 3. The pristine sample desorbs 4.1 wt% of H2 within the first 3 hours of measuring time (including the time for the heating ramp). Due to the additional weight of the additive, H2 capacity reduces to 3.2 wt% when 0.3 mol of potassium hydride is introduced. In order to compare the reaction rates, slopes between 20 and 70% (linear range of the dehydrogenation curves, Fig. 3a) of the normalized capacities are calculated (ESI Fig. 5). The reaction rate during the first dehydrogenation slightly increases with the addition of a tiny amount of KH (rate without additive: 1.7 reacted fraction unit/h and + 0.07 mol KH: 2.9 reacted fraction unit/h). Comparing the rates as more amount of KH is added, the difference in the reaction rate is not as significant (+ 0.15 mol KH: 2.87 reacted fraction unit/h, + 0.30 mol KH: 2.71 reacted fraction unit/h). After 25 H2 absorption/desorption cycles, the capacity remains stable for all additive-containing samples, while the H2 capacity drops about 50%, to 2.2 wt% for the pristine sample. Hence, the cycling temperature of 180 °C is not sufficient for the pristine sample to keep stable cycling properties. In order to compare the reaction rates at 25th desorption, slopes between 5 and 30% (linear range of the dehydrogenation curves, Fig. 3b) of the normalized capacities are calculated (ESI Fig. 6). Increasing the additive amount leads to an improvement in reaction rates (slope without additive: 0.88 reacted fraction unit/h, + 0.07 mol KH: 0.97 reacted fraction unit/h, + 0.15 mol KH:1.15 reacted fraction unit/h, + 0.30 mol KH: 2.13 reacted fraction unit/h). In summary, on the one hand, the presence of a tiny amount of KH speeds up the hydrogen release during the first cycle, but no further improvement is seen as the amount of KH increases. On the other hand, after 25 cycles, samples with a higher amount of KH initially (up to 30% of the reaction) show faster dehydrogenation kinetic behaviour. After the analysed linear range, the reaction rate behaviour of all material slows down significantly.
Structural characterization of the crystalline phases
Figure 4 shows the ex-situ XRD patterns of as-milled samples. The broad diffraction peak at Q = 15 nm−1 is related to the polymer material of the sample holder. None of the observed peaks are attributed to the Mg(NH2)2 phase, which tends to have amorphous nature after intense ball milling. Moreover, diffraction peaks of KH are also not visible at the sample with 0.07 mol of KH due to the low amount. However, KH is visible in the samples containing higher amounts. MgO is also observed at Q = 29 nm−1, suggesting the presence of some impurities, possibly during the synthesis of Mg(NH2)2.

XRD patterns of as-milled samples
In order to study the crystalline phase changes during the heating process, in-situ SR-PXD data was collected. Figure 5 shows the 2D contour plot of Mg(NH2)2 + LiH + 0.07KH sample, which contains the highest reversible capacity (4.2 wt%) and optimum reaction kinetics compared to other samples. Diffraction peaks of potassium hydride were not identified with ex-situ XRD (Fig. 4). Interestingly, during the first dehydrogenation (Fig. 5a), no diffraction peaks can match with any potassium-related compound. It is suggested that at this stage, K-containing phases are either amorphous or nanocrystalline. Once the temperature is reaching to more than 125 °C, diffraction peaks related to the main desorption phase, orthorhombic Li2Mg(NH)2 start to appear at Q = 21 nm−1 and 35 nm−1. Thereafter, the sample is cooled down to room temperature and loaded with 80 bar of H2 (Fig. 5b). Crystalline KH diffraction peaks appear at 125 °C at Q = 19 nm−1, together with the formation of broad peaks related to Mg(NH2)2 at Q = 10 nm−1. At this stage absorption takes place, however, magnesium amide is not crystalline until the temperature reaches 180 °C. At this temperature under isothermal conditions, when Mg(NH2)2 and LiH recrystallizes, no further change is observed. Amide-imide transformation is visible at the 2nd H2 desorption (Fig. 5c), but at higher temperatures (~ 125 °C at the 1st desorption, ~ 170 °C at the 2nd desorption).

In-situ XRD patterns of Mg(NH2)2 + 2LiH + 0.07KH sample: a 1st desorption, b 1st absorption, c 2nd desorption
Chemical state of the potassium-related phases
In the previous section, the overall sample composition and thermal behaviour were shown. However, the chemical state of potassium in the as-milled samples is not unknown due to the poor crystallinity of the samples after the high-energy ball-milling process. In order to observe the chemical state of potassium, XANES measurements at the potassium K-edge were performed (Fig. 6). XANES spectra of samples with 0.15 and 0.30 mol of KH indicate the presence of KH, which is in good agreement with results obtained from the ex-situ XRD patterns. However, the spectra are not fitting perfectly with the spectra of the reference KH, hence a potassium-containing phase with different chemical nature can be present. Interestingly, the XANES spectrum of the sample containing 0.07 mol of KH is not consistent with the spectra of the other two samples, meaning a different potassium compound could have been formed during the milling process. Indeed, the reaction of KH with Mg(NH2)2 during mechanical milling to form K2Mg(NH2)4 is highly favourable [17]. Although the spectrum is not identical to the spectrum of K2Mg(NH2)4, it is likely that some amount of KH reacts with Mg(NH2)2 during the ball-milling procedure to form K2Mg(NH2)4. On the contrary, Wang et al. [8] reported that only KH is present after the ball-milling procedure, and K2Mg(NH2)4 starts to form only during the dehydrogenation reaction. In the present work, the formation of K2Mg(NH2)4 after the ball-milling procedure can be related to the selection of higher milling speed (400 rpm instead of 200 rpm).

XANES spectra of as-milled samples at the absorption edge of potassium. Spectra for the reference powders, KH and K2Mg(NH2)4, are included in the graph
Structural evolution over cycling
Using small-angle X-ray scattering (SAXS), the nanostructure of a material can be investigated [27, 28]. In SAXS, the intensity of scattered X-rays is measured as a function of the scattering angle below 10°. In order to investigate the size evolution of the overall matrix structures over cycling, SAXS measurements on pristine and doped samples were carried out, and the results are shown in Fig. 7. The intensity axes in Fig. 7 are weighted by q2 in order to pronounce all features in the scattering intensities. The SAXS curve of the samples without additive (Mg(NH2)2 + 2LiH) exhibit a prominent feature in the Porod-region (at the very high q-values) and a broad shoulder in the Guinier-region (at the very low q-values), respectively. Upon cycling, the feature in the Porod-region becomes more pronounced but remains roughly stable on the q-axis at values around ~ 3 nm−1. These structures are about 2–6 nm in size. The feature visible in the Guinier-region (around 0.2 nm−1) of the as-milled sample shifts toward lower angles after the 1st cycle and migrates to even smaller q-values (out of the detectable q-range) after the 25th cycle. This implies a significant coarsening effect of the matrix structures in the pristine sample upon cycling. These two major structural features are also present in the as-milled sample with additive (Mg(NH2)2 + 2LiH + 0.07KH). The feature in the Porod-region is located roughly at the same q-values as in the pristine sample and the positions remain roughly unchanged over cycling. However, it becomes more prominent with cycling in comparison to the corresponding feature in the pristine sample. This indicates a higher contrast, due to the presence of the higher electronic number of potassium in comparison to the pristine sample. Similar to the pristine sample, a broad shoulder is present in the Guinier-region (at the very low q-values) of the as-milled sample, as well, which becomes much broader after the 1st desorption and remains roughly stable until the 25th cycle. This indicates a significantly lower coarsening effect in the matrix structures in the sample doped with KH over cycling relative to the pristine sample. Therefore, in comparison to the pristine sample, it is likely that the presence of potassium-containing phases at the amide-hydride interfaces stabilizes the particle sizes, and with this, the cycling stability by improving the mass transportation (shorter diffusion paths) at the interfaces.

Kratky plots for the samples with (Mg(NH2)2 + 2LiH + 0.07KH) and without additive (Mg(NH2)2 + 2LiH) at different cycle numbers
In order to obtain the size evolution of potassium-containing phases, ASAXS measurements were carried out (Fig. 8). To the best of our knowledge, ASAXS method at potassium K-edge is reported here for the 1st time for the amide-hydride systems. Since the K-absorption edge of potassium is at a low energy (~ 3.6 keV), attenuation of the X-rays by the sample is rather high. Hence, the application of ASAXS method at such low energies is rather challenging. Preparation of thin samples and rather long measuring times are required to obtain sufficient high-quality ASAXS curves. Resonant curves were obtained based on the difference between two ASAXS curves at different energies. Thereafter, they were fitted by assuming a spherical particle model. Figure 8 demonstrates that after ball milling of 36 h, potassium-containing nanoparticles with an average radius of 5 nm are present (See ESI Figs. 7–10 for fitting of scattering curves). After the 1st cycle, the average radius of these particles increases to ~ 7 nm, and after 25 cycles it further increases to ~ 10 nm. Although the presence of potassium-containing nanoparticles at the reaction interfaces improves the mass transportation (Fig. 7), significant growth of these particles may explain the degradation of hydrogen reaction kinetic properties of this system upon cycling (Fig. 3).

Size evolution of potassium-containing phases in the Mg(NH2)2 + 2LiH + 0.07KH sample over cycling. Data is obtained from the fitting of ASAXS curves (ESI Figures 7–10)
As seen previously from Fig. 7 and 8, particle sizes of both overall matrix and potassium-containing additive are growing over the cycling procedure. So far, both absorption and desorption cycles were carried out at 180 °C. In order to investigate the effect of temperature on the structure of the system, the absorption cycle temperature for the sample with 0.07 mol of additive is reduced to 150 °C (Fig. 9). SAXS curves of this sample at different cycling conditions illustrate clearly that the temperature has a great structural impact on the hydride matrix. When the sample is cycled at a lower absorption temperature, there are structures in the matrix with maximum diameters up to 100 nm (See ESI Figs. 11–12 for fitting of scattering curves).

Size distribution of Mg(NH2)2 + 2LiH + 0.07KH sample after 10 cycles. Data is obtained from the fitting of corresponding SAXS curves (ESI Figs. 11–12). Different colours are representing the difference in the absorption temperature during the cycling procedure
The higher absorption temperature results in an increment of the particle size diameters up to 400 nm. For comparison, the reaction kinetics of this sample at two different cycling conditions is measured (ESI Fig. 13). If the absorption temperature of 150 °C is chosen, 80% of total H2 capacity is released within 3.1 h. If the absorption temperature is set to 180 °C, the same capacity of H2 is released within 6.3 h. Therefore, the results clearly show that coarsening of the hydride matrix over the cycles is the main reason for the poor reaction kinetics. Furthermore, the results suggest that the coarsening effect can be minimized by lowering the operational absorption temperature. On the one hand, the cycling temperature may be reduced to minimize the growth of the particle sizes. On the other hand, further reducing the cycling temperature, especially the desorption temperature, may slow down the reaction kinetics, since lower temperatures mainly reduce the driving force for the dehydrogenation of metal hydrides.
Conclusion
The effect of KH on the Mg(NH2)2 + 2LiH hydrogen storage system is investigated by applying different laboratory and synchrotron characterization techniques in this work. Thermal analyses (Fig. 1) show that the addition of KH results in the reduction of the dehydrogenation onset temperature from 170 to 130 °C. However, the presence of KH also accounts for higher apparent activation energies (Table 1). In this regard, rate constants of the KH added materials measured under isothermal conditions of 180 °C show higher reaction rates than the pristine material. This phenomenon is attributed to the increased frequency factor ‘A’, indicating a more efficient interaction of the reactants. Volumetric studies (Fig. 3) show that by adding 0.07 mol of KH, stable hydrogen capacity (4.2 wt%) over the first 25 cycles can be obtained. Presence of a tiny amount of KH speeds up the hydrogen release during the 1st cycle, but no further improvement is seen as the amount of KH increases (Fig. 3a). After 25 cycles, samples with a higher amount of KH initially (up to 30% of the reaction) show faster dehydrogenation kinetic behaviour (Fig. 3b). Later on, the reaction rate behaviour of all material slows down significantly. Potassium-containing phases, possibly at the amide-hydride interfaces, stabilize the overall particle size and suppress growth, compared to the particle growth of the pristine sample (Fig. 7). Thus, such potassium-containing phases shorten diffusion paths and ensure full reversibility of the reaction, improving the overall cycling stability. However, the average particle size of potassium-containing phases still grows after 25 cycles (Fig. 8), which explains the partial degradation of hydrogen reaction kinetic properties of this system upon cycling (Fig. 3). The degradation effect can be minimized by reducing the absorption temperature from 180 to 150 °C, which prevents the growth of nanoparticles (Fig. 9). As a consequence, 2× faster reaction kinetics can be obtained (ESI Fig. 13). All in all, the information obtained from this work can be useful to design and develop reversible hydrogen storage systems for stationary and mobile applications.
References
Eberle U, Felderhoff M, Schüth F (2009) Chemische und physikalische lösungen für die speicherung von wasserstoff. Angew Chemie 121:6732–6757. https://doi.org/10.1002/ange.200806293
Chen P, Xiong Z, Luo J, Lin J, Lee Tan K (2002) Interaction of hydrogen with metal nitrides and imides. Nature 420(6913):302–304. https://doi.org/10.1038/nature01210
Wang J, Li HW, Chen P (2013) Amides and borohydrides for high-capacity solid-state hydrogen storage-materials design and kinetic improvements. MRS Bull 38(6):480–487. https://doi.org/10.1557/mrs.2013.131
Xiong Z, Wu G, Hu J, Chen P (2004) Ternary imides for hydrogen storage. Adv Mater 16(17):1522–1525. https://doi.org/10.1002/adma.200400571
Luo W, Sickafoose S (2006) Thermodynamic and structural characterization of the Mg–Li–N–H hydrogen storage system. J Alloys Compd 407(1–2):274–281. https://doi.org/10.1016/j.jallcom.2005.06.046
Luo W (2004) (LiNH2–MgH2): a viable hydrogen storage system. J Alloys Compd. https://doi.org/10.1016/j.jallcom.2004.03.119
Ropp RC (2003) Solid state chemistry. Elsevier, Amsterdam, Chapter 4
Wang J et al (2009) Potassium-modified Mg(NH2)2/2LiH system for hydrogen storage. Angew Chemie Int Ed 48(32):5828–5832. https://doi.org/10.1002/anie.200805264
Durojaiye T, Hayes J, Goudy A (2015) Potassium, rubidium and cesium hydrides as dehydrogenation catalysts for the lithium amide/magnesium hydride system. Int J Hydrog Energy 40(5):2266–2273. https://doi.org/10.1016/j.ijhydene.2014.12.056
Cao H, Wu G, Zhang Y, Xiong Z, Qiu J, Chen P (2014) Effective thermodynamic alteration to Mg(NH2)2–LiH system: achieving near ambient-temperature hydrogen storage. J Mater Chem A 2(38):15816–15822. https://doi.org/10.1039/c4ta03505d
Gizer G et al (2019) Enhancement effect of bimetallic amide K2Mn(NH2)4 and in-situ formed KH and Mn4N on the dehydrogenation/hydrogenation properties of Li–Mg–N–H system. Energies 12(14):2779. https://doi.org/10.3390/en12142779
Liang C et al (2011) Enhanced dehydrogenation/hydrogenation kinetics of the Mg(NH2)2–2LiH system with NaOH additive. Int J Hydrog Energy 36(3):2137–2144. https://doi.org/10.1016/j.ijhydene.2010.11.068
Shahi RR, Yadav TP, Shaz MA, Srivastva ON (2010) Studies on dehydrogenation characteristic of Mg(NH2)2/LiH mixture admixed with vanadium and vanadium based catalysts (V, V2O5 and VCl3). Int J Hydrog Energy 35(1):238–246. https://doi.org/10.1016/j.ijhydene.2009.10.029
Bill RF, Reed D, Book D, Anderson PA (2015) Effect of the calcium halides, CaCl2 and CaBr 2, on hydrogen desorption in the Li–Mg–N–H system. J Alloys Compd 645:S96–S99. https://doi.org/10.1016/j.jallcom.2014.12.269
Hu J, Weidner E, Hoelzel M, Fichtner M (2010) Functions of LiBH4 in the hydrogen sorption reactions of the 2LiH–Mg(NH2)2 system. Dalton Trans 39(38):9100. https://doi.org/10.1039/c0dt00468e
Wang J et al (2009) Potassium-modified Mg(NH2)2/2LiH system for hydrogen storage. Angew Chemie Int Ed. https://doi.org/10.1002/anie.200805264
Wang J et al (2013) Solid–solid heterogeneous catalysis: the role of potassium in promoting the dehydrogenation of the Mg(NH2)2/2LiH composite. Chemsuschem 6(11):2181–2189. https://doi.org/10.1002/cssc.201200885
Li C, Liu Y, Yang Y, Gao M, Pan H (2014) High-temperature failure behaviour and mechanism of K-based additives in Li–Mg–N–H hydrogen storage systems. J Mater Chem A 2(20):7345–7353. https://doi.org/10.1039/C4TA00025K
Cerenius Y et al (2000) The crystallography beamline I711 at MAX II. J Synchrotron Radiat 7(4):203–208. https://doi.org/10.1107/S0909049500005331
Bösenberg U et al (2014) Characterization of metal hydrides by in-situ XRD. Int J Hydrog Energy. https://doi.org/10.1016/j.ijhydene.2014.02.068
Pistidda C et al (2015) First direct study of the ammonolysis reaction in the most common alkaline and alkaline earth metal hydrides by in situ SR-PXD. J Phys Chem C. https://doi.org/10.1021/jp510720x
Rietveld HM (1968) A profile refinement method for nuclear and magnetic structures. J Appl Crystallogr 2:65–71. https://doi.org/10.1107/s0021889869006558
Krumrey M (1998) Design of a four-crystal monochromator beamline for radiometry at BESSY II. J Synchrotron Radiat. https://doi.org/10.1107/S0909049597011825
Wernecke J, Gollwitzer C, Müller P, Krumrey M (2014) Characterization of an in-vacuum PILATUS 1M detector. J Synchrotron Radiat 21(3):529–536. https://doi.org/10.1107/S160057751400294X
Haas S et al (2010) Direct accessing the nanostructure of carbon supported Ru–Se based catalysts by ASAXS. J Phys Chem C 114(51):22375–22384. https://doi.org/10.1021/jp103340t
Gericke E et al (2021) Direct observation of the xenon physisorption process in mesopores by combining in situ anomalous small-angle X-ray scattering and X-ray absorption spectroscopy. J Phys Chem Lett 12(16):4018–4023. https://doi.org/10.1021/acs.jpclett.1c00557
Karimi F et al (2021) A comprehensive study on lithium-based reactive hydride composite (Li-RHC) as a reversible solid-state hydrogen storage system toward potential mobile applications. RSC Adv 11(37):23122–23135. https://doi.org/10.1039/d1ra03246a
Karimi F et al (2021) Characterization of LiBH4–MgH2 reactive hydride composite system with scattering and imaging methods using neutron and synchrotron radiation. Adv Eng Mater. https://doi.org/10.1002/adem.202100294
Acknowledgements
The authors gratefully acknowledge usage of the PTB FCM-beamline, and the support of the PTB Staff, Michael Krumrey and Levent Cibik. We thank the Helmholtz-Zentrum Berlin für Materialien und Energie for the allocation of synchrotron radiation beamtime.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no competing financial interest.
Additional information
Handling Editor: M. Grant Norton.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gizer, G., Karimi, F., Pistidda, C. et al. Effect of the particle size evolution on the hydrogen storage performance of KH doped Mg(NH2)2 + 2LiH. J Mater Sci 57, 10028–10038 (2022). https://doi.org/10.1007/s10853-022-06985-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-06985-4