
Abstract
The concept of chemical space is playing an increasingly important role in many areas of chemical research, especially medicinal chemistry and chemical biology. It is generally conceived as consisting of numerous compound clusters of varying sizes scattered throughout the space in much the same way as galaxies of stars inhabit our universe. A number of issues associated with this coordinate-based representation are discussed. Not the least of which is the continuous nature of the space, a feature not entirely compatible with the inherently discrete nature of chemical space. Cell-based representations, which are derived from coordinate-based spaces, have also been developed that facilitate a number of chemical informatic activities (e.g., diverse subset selection, filling ‘diversity voids’, and comparing compound collections).These representations generally suffer the ‘curse of dimensionality’. In this work, networks are proposed as an attractive paradigm for representing chemical space since they circumvent many of the issues associated with coordinate- and cell-based representations, including the curse of dimensionality. In addition, their relational structure is entirely compatible with the intrinsic nature of chemical space. A description of the features of these chemical space networks is presented that emphasizes their statistical characteristics and indicates how they are related to various types of network topologies that exhibit random, scale-free, and/or ‘small world’ properties.


Similar content being viewed by others
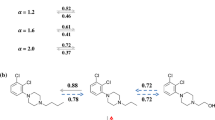


Notes
When all vertices are connected to each other the network is called complete. Complete undirected networks with n vertices have n (n − 1)/2 edges.
Similarity values that are strictly greater than a given threshold are called proper threshold values.
Note that in rare cases the similarity between two dissimilar molecules may, nonetheless, be unity. This is due to limitations of the molecular representation that do not properly account for all the features that distinguish the two molecules from one another.
\({\text{SALI}}(i,j) = {{\left| {\varDelta {\text{Act}}(i,j)} \right|} \mathord{\left/ {\vphantom {{\left| {\varDelta {\text{Act}}(i,j)} \right|} {\left[ {1 - {\text{Sim}}(i,j)} \right]}}} \right. \kern-0pt} {\left[ {1 - {\text{Sim}}(i,j)} \right]}}\), where ∆Act(i, j) is the difference in the activities of a given compound pair (i, j), and Sim(i, j) is their corresponding the similarity value.
In the case of directed graphs, the degree of incoming connections (‘in-degree’) is considered separately for that of the outgoing connections (‘out-degree’).
Hence, shortest paths are integer valued. Note that it is possible that more than one shortest path exists between a given pair of vertices.
References
Workshop on Navigating chemical compound space for materials and bio design, held at the Institute for Pure and Applied Mathematics, University of California, Los Angeles, CA, March 14–June 17, 2011. https://www.ipam.ucla.edu/programs/ccs2011/. Accessed 3 April 2014
Dobson CM (2004) Chemical space and biology. Nature 432:824–828
Bellman RE (1961) Adaptive control processes. Princeton University Press, Princeton
Hecht-Nielsen R (1990) Neurocomputing. Addison-Wesley Publishing Company, Reading
Pearlman R, Smith K (2002) Novel software tools for chemical diversity. 3D QSAR Drug Design 2:339–353
Barabási A-L (2003) Linked—how everything is connected to everything else and what it means for business, science, and everyday life. PLUME, Penguin Books, New York
Watts DJ (2003) Six degrees—the science of a connected age. W.W. Norton & Company, New York
Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL (2006) Global mapping of pharmacological space. Nat Biotechnol 24:805–815
Hopkins AL (2008) Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4:682–690
Keiser MJ, Roth BL, Armruster BN, Ernsberger P, Irwin JJ, Shoichet BK (2007) Relating protein pharmacology by ligand chemistry. Nat Biotechnol 25:197–206
Yildirim MA, Goh K-I, Cusick ME, Barabási A-L, Vidal M (2007) Drug-target network. Nat Biotechnol 25:1119–1126
Tanaka N, Ohno K, Niimi T, Moritomo A, Mori K, Orita M (2009) Small-world phenomena in chemical library networks: application to fragment-based drug discovery. J Chem Inf Model 49:2677–2686
Krein MP, Sukumar N (2011) Exploration of the topology of chemical spaces with network measures. J Phys Chem A 115:12905–12918
Wawer M, Peltason L, Weskamp N, Teckentrup A, Bajorath J (2008) Structure-activity relationship anatomy by network-like similarity graphs and local structure-activity relationship indices. J Med Chem 51:6075–6084
Ripphausen P, Nisius B, Wawer M, Bajorath J (2011) Rationalizing the role of SAR tolerance for ligand-based virtual screening. J Chem Inf Model 51:837–842
Stumpfe D, Dimova D, Bajorath J (2014) Composition and topology of activity cliff clusters formed by bioactive compounds. J Chem Inf Model 54:451–461
Cohen R, Havlin S (2009) Scaling properties of complex networks and spanning trees. In: Bollobás B, Kozma R, Miklós (eds) Handbook of large-scale random networks. Springer, New York, pp 143–169
Newman MEJ (2010) Networks—an introduction. Oxford University Press Inc., New York
Wasserman S, Faust K (1994) Social network analysis—methods and applications. Cambridge University Press, Cambridge
Guha R, Van Drie JH (2008) Structure-activity landscape index: identifying and quantifying activity cliffs. J Chem Inf Model 48:646–658
Jain AK, Dubes RC (1988) Algorithms for clustering data. Prentice-Hall, Engelwood Cliffs
Kolaczyk ED (2009) Statistical analysis of network data—methods and models. Springer, New York
Albert R, Barabási A-L (2002) Statistical mechanics of complex networks. Rev Mod Phys 74:47–97
Barabási A-L, Albert R (1999) Emergence of scaling in random networks. Science 286:509–512
Watts DJ (1999) Small worlds—the dynamics of networks between order and randomness. Princeton University Press, Princeton
Watts DJ, Strogatz SH (1998) Collective dynamics of ‘small world’ networks. Nature 393:440–442
Benz RW, Swamidass SJ, Baldi P (2008) Discovery of power-laws in chemical space. J Chem Inf Model 48:1138–1151
Schneider G, Neidhart W, Giller T, Schmid G (1999) ‘Scaffold hopping’ by topological pharmacophore search: a contribution to virtual screening. Angew Chem Int Ed 38:2894–2896
Iyer P, Stumpfe D, Vogt M, Bajorath J, Maggiora GM (2013) Activity landscapes, information theory, and structure-activity relationships. Mol Inf 32:421–430
Birchall K, Gillet VJ (2011) Reduced graphs and their applications in chemoinformatics, chapter 8. In: Bajorath J (ed) Chemoinformatics and computational chemical biology. Springer, New York, pp 197–212
Liu B (2011) Web data mining: exploring hyperlinks, contents, and usage data. Springer, Heidelberg
Robinson I, Webber J, Elfrem E (2013) Graph databases. O’Reilly Media Inc., Sebastopol
Acknowledgments
The authors wish to thank Dr. Vijay Gokhale for reading the manuscript and for his helpful comments and Dr. Dagmar Stumpfe for the design of exemplary network representations and review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maggiora, G.M., Bajorath, J. Chemical space networks: a powerful new paradigm for the description of chemical space. J Comput Aided Mol Des 28, 795–802 (2014). https://doi.org/10.1007/s10822-014-9760-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-014-9760-0