
Abstract
Rett syndrome (RTT) is a rare monogenic disorder affecting 1 in 10,000 live female births causing severe neurodegenerative symptoms. We analyzed the molecular genetic variants in the gene encoding the methyl-CpG binding protein 2 (MECP2) of 16 girls with RTT. Their mutation profile was as follows; Already described variants: p.R168X in 25% (n = 4), p.T158M in 25% (n = 4), p.R255X in 12.5% (n = 2), p.R133C in 12.5% (n = 2), p.R294X in 6.25% (n = 1), p.K177X in 6.25% (n = 1). Novel variants: a large deletion (c.868_1188del321) in 6.25% (n = 1) and a p.X499L in 6.25% (n = 1). We also looked at the genotype to phenotype correlation of these variants. Most of the mutations were C>T in CpG hot spot as seen in other populations.



Similar content being viewed by others

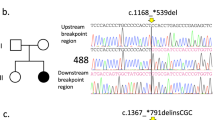
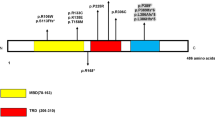
Data Availability
Availability of data and materials—all data is published in the manuscript material is available with the authors. We, however, can’t provide personal information or data containing identification of patients in any form.
Abbreviations
- RTT:
-
Rett syndrome
- MECP2:
-
Methyl‐CpG‐binding protein 2
- MBD:
-
Methyl-CpG binding domain
References
Adzhubei, I., Schmidt, S., Peshkin, L., Ramensky, V., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nature Methods,7(4), 248–249.
Amir, R., Van den Veyver, I., Wan, M., Tran, C., Francke, U., & Zoghbi, H. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics,23(2), 185–188.
Archer, H. (2006). CDKL5 mutations cause infantile spasms, early-onset seizures, and severe mental retardation in female patients. Journal of Medical Genetics,43(9), 729–734.
Ariani, F., Hayek, G., Rondinella, D., Artuso, R., Mencarelli, M., Spanhol-Rosseto, A., et al. (2008). FOXG1 is responsible for the congenital variant of Rett syndrome. The American Journal of Human Genetics,83(1), 89–93.
Baker, S., Chen, L., Wilkins, A., Yu, P., Lichtarge, O., & Zoghbi, H. (2013). An AT-Hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell,152(5), 984–996.
Bebbington, A., Anderson, A., Ravine, D., Fyfe, S., Pineda, M., de Klerk, N., et al. (2008). Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology,70(11), 868–875.
Beyer, K., Blasi, F., Bacchelli, E., Klauck, S., Maestrini, E., Poustka, A., et al. (2002). Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Human Genetics,111(4–5), 305–309.
Buschdorf, J., & Stratling, W. (2004). A WW domain binding region in methyl-CpG-binding protein MeCP2: Impact on Rett syndrome. Journal of Molecular Medicine,82(2), 135–143.
Calfa, G., Percy, A., & Pozzo-Miller, L. (2011). Experimental models of Rett syndrome based on Mecp2 dysfunction. Experimental Biology and Medicine,236(1), 3–19.
Chahrour, M., Jung, S., Shaw, C., Zhou, X., Wong, S., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science,320(5880), 1224–1229.
Chandler, S., Guschin, D., Landsberger, N., & Wolffe, A. (1999). The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry,38(22), 7008–7018.
Charman, T., Neilson, T., Mash, V., Archer, H., Gardiner, M., Knudsen, G., et al. (2005). Dimensional phenotypic analysis and functional categorization of mutations reveal novel genotype-phenotype associations in Rett syndrome. European Journal of Human Genetics,13(10), 1121–1130.
Cheadle, J. (2000). Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Human Molecular Genetics,9(7), 1119–1129.
Cuddapah, V., Pillai, R., Shekar, K., Lane, J., Motil, K., Skinner, S., et al. (2014). Methyl-CpG-binding protein 2(MECP2) mutation type is associated with disease severity in Rett syndrome. Journal of Medical Genetics,51(3), 152–158.
D’Esposito, M., Quaderi, N., Ciccodicola, A., Bruni, P., Esposito, T., D’Urso, M., et al. (1996). Isolation, physical mapping, and Northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mammalian Genome,7(7), 533–535.
Girard, M., Couvert, P., Carrié, A., Tardieu, M., Chelly, J., Beldjord, C., et al. (2001). Parental origin of de novo MECP2 mutations in Rett syndrome. European Journal of Human Genetics,9(3), 231.
Glaze, D. G., Percy, A. K., Skinner, S., Motil, K. J., Neul, J. L., Barrish, J. O., et al. (2010). Epilepsy and the natural history of Rett syndrome. Neurology,74(11), 909–912.
Guy, J., Alexander-Howden, B., FitzPatrick, L., DeSousa, D., Koerner, M. V., Selfridge, J., et al. (2018). A mutation-led search for novel functional domains in MeCP2. Human Molecular Genetics,27(14), 2531–2545.
Hagberg, B., Aicardi, J., Dias, K., & Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Annals of Neurology,14(4), 471–479.
Hoffbuhr, K., Devaney, J. M., LaFleur, B., Sirianni, N., Scacheri, C., Giron, J., et al. (2001). MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology,56(11), 1486–1495.
Horvath, P. M., & Monteggia, L. M. (2018). MeCP2 is an activator of gene expression. Trends in Neurosciences,41(2), 72–74.
Ip, J. P., Mellios, N., & Sur, M. (2018). Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nature Reviews Neuroscience,19(6), 368.
Jian, L., Nagarajan, L., de Klerk, N., Ravine, D., Christodoulou, J., & Leonard, H. (2007). Seizures in Rett syndrome: An overview from a one-year calendar study. European Journal of Paediatric Neurology,11(5), 310–317.
Lallar, M., Rai, A., Srivastava, P., Mandal, K., Gupta, N., Kabra, M., et al. (2018). Molecular testing of MECP2 gene in Rett syndrome phenotypes in Indian girls. Indian Pediatrics,55(6), 474–477.
Larvick, C. L., de Klerk, N., Bower, C., Christodoulou, J., Ravine, D., Ellaway, C., et al. (2006). Rett syndrome in Australia: A review of the epidemiology. The Journal of pediatrics,148(3), 347–352.
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell,69(6), 905–914.
Li, M. R., Pan, H., Bao, X. H., Zhang, Y. Z., & Wu, X. R. (2007). MECP2 and CDKL5 gene mutation analysis in Chinese patients with Rett syndrome. Journal of Human Genetics,52(1), 38.
Liu, K., Xu, C., Lei, M., Yang, A., Loppnau, P., Hughes, T. R., et al. (2018). Structural basis for the ability of MBD domains to bind methyl-CG and TG sites in DNA. Journal of Biological Chemistry,293(19), 7344–7354.
Lopes, F., Barbosa, M., Ameur, A., Soares, G., de Sá, J., Dias, A. I., et al. (2016). Identification of novel genetic causes of Rett syndrome-like phenotypes. Journal of Medical Genetics, 53(3), 190–199.
Müller, M. (2019). Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neuroscience & Biobehavioral Reviews.
Neul, J. L., Fang, P., Barrish, J., Lane, J., Caeg, E. B., Smith, E. O., et al. (2008). Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology,70(16), 1313–1321.
Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi‐Buisson, N., … & Renieri, A. (2010). Rett syndrome: Revised diagnostic criteria and nomenclature. Annals of neurology, 68(6), pp. 944–950
Percy, A. K. (2008). Rett syndrome: Recent research progress. Journal of child neurology,23(5), 543–549.
Percy, A. K., Lane, J. B., Childers, J., Skinner, S., Annese, F., Barrish, J., et al. (2007). Rett syndrome: North American database. Journal of Child Neurology,22(12), 1338–1341.
Psoni, S., Sofocleous, C., Traeger-Synodinos, J., Kitsiou-Tzeli, S., Kanavakis, E., & Fryssira-Kanioura, H. (2012). MECP2 mutations and clinical correlations in Greek children with Rett syndrome and associated neurodevelopmental disorders. Brain and Development,34(6), 487–495.
Rai, V. (2011). Autism genetics: Recent advances in candidate gene search. International J Genetic Engineering and Biotechnology,2(1), 47–66.
Schanen, C., Houwink, E. J., Dorrani, N., Lane, J., Everett, R., Feng, A., et al. (2004). Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. American Journal of Medical Genetics Part A,126(2), 129–140.
Schwarz, J. M., Cooper, D. N., Schuelke, M., & Seelow, D. (2014). Mutation Taster2: Mutation prediction for the deep-sequencing age. Nature Methods,11(4), 361.
Steffenburg, U., Hagberg, G., & Hagberg, B. (2001). Epilepsy in a representative series of Rett syndrome. Acta Paediatrica,90(1), 34–39.
Suter, B., Treadwell-Dearing, D., Zoghbi, H., Glaze, D., & Neul, J. L. (2010). MECP2 mutations in people without Rett syndrome. Annals of Neurology,68(4), S98–S99.
Townend, G. S., Ehrhart, F., van Kranen, H. J., Wilkinson, M., Jacobsen, A., Roos, M., et al. (2018). MECP2 variation in Rett syndrome—An overview of current coverage of genetic and phenotype data within existing databases. Human Mutation,39(7), 914–924.
Trappe, R., Laccone, F., Cobilanschi, J., Meins, M., Huppke, P., Hanefeld, F., et al. (2001). MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. The American Journal of Human Genetics,68(5), 1093–1101.
Weaving, L. S., Ellaway, C. J., Gecz, J., & Christodoulou, J. (2005). Rett syndrome: Clinical review and genetic update. Journal of Medical Genetics,42(1), 1–7.
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proceedings of the National Academy of Sciences,102(49), 17551–17558.
Zhu, X., Li, M., Pan, H., Bao, X., Zhang, J., & Wu, X. (2010). Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China. Journal of Child Neurology,25(7), 842–848.
Zoghbi, H. Y. (2003). Postnatal neurodevelopmental disorders: Meeting at the synapse? Science,302(5646), 826–830.
Acknowledgments
The authors wish to thank the laboratory staff the referring physicians and clinical geneticists. Special thanks to Dr. P.D. Ratnayake, Neurology Unit, Lady Ridgeway Hospital, Colombo, Sri Lanka and Dr. S.M.A. Jayawardena, Faculty of Medicine and Allied Sciences, Rajarata University of Sri Lanka. Authors wish to thank the laboratory staff of the Asiri Sugical Hospital and the Human Genetics Unit faculty of Medicine University of Colombo and the referring physicians.
Funding
There was no funding for this study.
Author information
Authors and Affiliations
Contributions
DH wrote the first draft of the manuscript with contributions from all. All authors reviewed, modified and approved the final version of the manuscript. NFN and BAPSP performed the analysis. VHWD designed and guided the study.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no competing interests.
Ethics Approval
Ethics approval was obtained by the ERC of The Faculty of Medicine University of Colombo to study rare genetic conditions in the Sri Lankan population. The procedure was part of the patient’s care and posed no additional risk to the patients’ health. No identifiable information of the study participants is disclosed. All procedures performed in studies involving human participants were by the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from study participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hettiarachchi, D., Neththikumara, N.F., Pathirana, B.A.P.S. et al. Variant Profile of MECP2 Gene in Sri Lankan Patients with Rett Syndrome. J Autism Dev Disord 50, 118–126 (2020). https://doi.org/10.1007/s10803-019-04230-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-019-04230-7