
Abstract
Simple and rapid analysis of cadmium ion in environmental and biological samples is of great importance due to the severe toxicity caused by this heavy metal. In the present work, nickel tungstate (NiWO4) dispersion was mixed with multi-walled carbon nanotubes (MWCNTs) to obtain a homogenous composite of (NiWO4/MWCNTs) which was assigned as carbon paste electrode modifier. The composite was fully characterized using various characterization techniques including X-Ray Diffraction (XRD), Scanning Electron Microscope (SEM), Thermo-Gravimetric Analysis (TGA) and Fourier Transform InfraRed (FTIR). The electrochemical redox reactions of cadmium (II) ions at the modified electrode interface were investigated using cyclic voltammetry (CV), differential pulse voltammetry (DPV), and linear sweep voltammetry (LSV). Effective parameters on the electro-analysis assay performance including the electrode composition, types of electrolyte, scan rate and pH were tested to achieve the best effective optimum conditions. Accordingly, a linear relation of cadmium ions was achieved in the concentration range 50–450 µM with limit of detection of 0.12 µM. Besides, the proposed electrode was successfully used to monitor trace amounts of cadmium ions in various real samples.
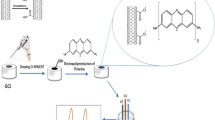
Graphical abstract
Schematic illustration of synthesis process of NiWO4/MWCN nanocomposite and its application as high-performance cadmium ion sensors

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Heavy metal ions are a major class of pollutants that have a significant impact on different aspects of the environment, including both terrestrial and aquatic ecosystems [1]. The increasing industrial activity has resulted in the freeing of heavy metals into the environment, in addition to the use of pesticides that contain metallic constituents led to the bioaccumulation of heavy metals in plants and animals that are part of the food chain [2]. Even small amounts of these heavy metal ions including cadmium, chromium, lead, zinc, copper, arsenic, and mercury can be extremely poisonous. Cadmium, in particular, is one of the most hazardous heavy metals and can cause various health problems, including major fatal conditions such as cancer, heart disease, and diabetes [3]. Cadmium is widely utilized in various industrial workplaces such as electroplating, metallurgy, and nickel–cadmium batteries. The usage of cadmium in these workplaces can cause significant harm to living organisms [4]. Therefore, it is necessary to keep tracking the level of cadmium concentration using quick, simple, sensitive, selective and efficient analytical methods.
In this regard, various analytical methods were developed and applied for the determination of cadmium including inductively coupled plasma atomic emission spectrometry (ICP-AES), inductively coupled plasma-mass spectrometry (ICP-MS) [5], atomic absorption spectroscopy (AAS) [6], and ion chromatography [7]. Although these methods have achieved high sensitivity, and low limit of detection, they are time consuming and costly. In addition, these methods do not meet portable demands, onsite detection, and consumption of small amounts of chemical reagents. Alternatively, electro-analytical methods alongside the electrochemical sensors and biosensors came to satisfy these expectancies and are suggested as best substitutional because of their ease of operation, fast analysis, low cost, portability, high selectivity, and superior sensitivity [8,9,10,11,12]. Materials and active surface area of working electrodes are necessary for regulating the redox reaction(s), enhancing the charge transfer as well as for supporting the analyte(s) deposition and accumulation [13, 14]. The common use of bare (unmodified) carbon electrodes in the electro-analysis is attributed to its low cost, low chemical reactivity, and simple preparation [15]. Adams et al. introduced carbon paste electrodes (CPEs) for the first time in 1958, and since that time, CPEs are strongly involved in many electro-analytical applications [14, 16]. Several important features demonstrated the popularity of CPEs (e.g. large potential window, low ohmic resistance, the simple surface functionalization and in-situ modification [17]. However, the use of unmodified CPEs provide low sensitivity (high detection limit) and low selectivity (interference problems). Thus, surface modification of CPEs, through the electro-deposition, drop-casting, or bulk modification, was taken into consideration to overcome those problems. In this regard, semiconductor NiWO4 was selected in this study with a narrow band gap of 2.2 eV and showed notable enhancement in electrical conductivity due to the presence of various valence states provoked by the incorporated tungsten oxides particles [18]. Thus, the incorporation of tungsten oxides particles will increase the conductivity and the catalytic properties of the NiWO4 in comparison with the pure NiO. Nickel tungstate (NiWO4) has higher electrical conductivity (10−7–10−2 Scm−1) than other multivalent binary oxides (NiMO4 ~ 10−11–10−4.5 Scm−1) [19] or many other transition metal oxides (FeWO4 ~ 10−6 Scm−1 [20], and Bi2WO6 ~ 10−8 Scm−1 [21]). Nickel tungstate has low fabrication cost, low rate of corrosion, high stability and high electro-catalytic activity. Further, it is environmentally friendly and has been utilized in a wide range of applications (e.g. lithium-ion batteries, super-capacitors, and electro-catalysis) [22]. Therefore, the sensing material (NiWO4) was applied as a promising electrode modifier to promote electron transfer kinetics and increase the adsorption efficiency in respect of Cd(II) onto the electrode surface.
In the present work, carbon paste electrode (CPE) was modified with a homogeneous dispersion of (NiWO4/MWCNTs) to enable high selectivity and sensitivity towards the voltammetric detection of cadmium ions in environmental and biological samples.
2 Experimental
2.1 Chemical reagents and substances
Nickel chloride hexa-hydrate (NiCl2⋅6H2O 99.9%), sodium tungstate di-hydrate (Na2WO4⋅2H2O ≥ 99%) were provided from Sigma–Aldrich. N,N-dimethylformamide (DMF ≥ 99.8%) was bought from Fisher. Parent solution of 1.0mM of cadmium was performed using cadmium nitrate (Cd(NO3)2 >98%) from Merck, Germany. Phthalic acid (98%) and potassium ferricyanide (FCN 98%), which were used as a supporting electrolyte and the standard redox probe, were obtained from the LOBA-CHEMIE-PVT. Multi-walled carbon nanotubes (MWCNTs, Sigma, Aldrich). Graphite powder and paraffin oil were provided by Aldrich, and Fluka, respectively. Double-distilled water was used as solvent for the preparation of all previous solutions.
3 Preparation of NiWO4 particles
According to the previously published method [23], nickel tungstate (NiWO4) was synthesized as follows: 0.1 M of sodium tungstate was gradually added to 0.1 M of nickel chloride solution (ration of 1:1 V/V) with continuous stirring at 70 oC. Solid precipitate was produced, and left in mother liquor for 24 h. Subsequently, the precipitate was thoroughly washed with double-distilled water to eliminate the remaining unreacted reagents, and lastly was filtered out using a suction pump. The product was dried in an oven at 70 ± 2 oC. The final product was suspended in double-distilled water to form a homogenous dispersion.
3.1 Modification of the working electrode (CPEs)
Modified carbon paste electrodes were prepared by hand mixing 500 mg of synthetic graphite powder with 75 µL paraffin oil in a ceramic mortar for 15 min. Then the homogenous carbon pastes were packed into cylindrical Teflon piston holders with internal diameter of 2.5 and 30 mm height and conductive electric screws for electric contact with the cables of the potentiostat. The electrode surface was smoothed on a wet-filter paper. To modify the surface of the electrodes, 10µL of the composite suspension contains (2.0 mg/mL of MWCNTs + 6 mg/mL of the NiWO4 in DMF) was placed by the drop casting method and left to dry at room temperature. The superficies of the modified electrodes were washed with water prior insertion into the electrochemical cell.
3.2 Instruments
Voltammetric studies were performed using potentiostat/galvanostat(AUTOLAB PGSTAT 302 N, Metrohm, Utrecht, Netherlands), connected with a three-electrode system including NiWO4/MWCNTs based carbon paste electrode as an active electrode, Pt disc as a counter electrode and a Ag/AgCl/3 M KCl reference electrode. Buffer pH values were adjusted with a pH-meter (JENWAY, Model 3510, UK). The FTIR spectra were collected using Bruker Vertex 70. Field emission Scanning Electron Microscope and element mapping FSEM/EDX were performed using (Gemini Zeiss-Sigma 500 VP). Thermal analysis tests viz., thermo-gravimetric analysis (TGA) and DTG were carried out using (DTG-60 H, Shimadzu, Japan). X-ray diffraction (XRD) patterns were obtained by (Shimadzu, Kyoto, Japan).
3.3 Electrochemical procedures
Before running the electrochemical measurement, the modified electrodes were activated in phthalic acid by running 10 voltammetric cyclic scans from − 0.9 to − 0.3 V with scan rate of 50 mV/s. Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and differential pulse voltammetry (DPV) were used for characterization, assay optimization and for quantitative analysis. The measurements were performed in a 30 mL electrochemical cell containing phthalic acid (pH 3.5) as the supporting electrolyte. The modified CPE was flooded in the electrolyte solution containing cadmium ions and then washed for 30 s to collect the metal ions around the electrode interface. Standard addition method was applied for generating calibration curves.
3.4 Real samples pre-treatment
Four different real water samples (tap water, River-Nile water, ground water, wastewater) and biological samples (urine samples) were collected and pre-treated before determination of Cd(II) ions in their contents. The collected samples were acidified and their pH was adjusted to pH = 2.0 with nitric acid. Spiked urine sample was acid-digested by adding 6.0 mL of concentrated nitric acid to 3.0 mL of the sample and heating the mixture until complete evaporation to eliminate the organic substances. Then the residue was solubilized by the addition of deionized water.
4 Results and discussion
4.1 Characterization of the chemically synthesized NiWO4
NiWO4 nanomaterial was characterized by a series of physical and structural analysis techniques such as FTIR, XRD, TGA/DTG, SEM, and CV.
The FTIR of the NiWO4 (Fig. 1a) was used to investigate its functional groups content. The bands appearing at wavenumbers of ~ 854 and 684 cm−1 are attributed to the existence of WO4 −2 and the groups of metal oxide, respectively [24], which confirms the formation of NiWO4. Besides, the band perceived at 3352 cm−1 is attributed to moisture in the sample and OH-group [25]. The strong stretching band observed at 1634 cm−1 is assigned to the vibration process of the free molecules of water. Whereas, the crest at 1386 cm−1 corresponds to the presence of vibration of hydroxyl group of water. Therefore, the obtained FTIR spectrum (Fig. 1a) confirmed the formation of NiWO4.

a FTIR spectra of NiWO4 nanoparticles, b XRD patterns of NiWO4, c TGA/DTG curves
The crystalline structure of the NiWO4 was elucidated using XRD analysis. In Fig. 1b, a pair of wide deviation crests with low intensity demonstrated the formation of tiny sizes of the NiWO4 with non-crystalline assembly. The diffraction patterns exhibited no crystalline phase before the calcination. This finding is in accordance with that has been reported in other previous studies [26]. Two broad peaks (appeared at 2θ = 33.56° and 59.73°) are attributed to (111) and (202) planes of NiWO4 (JCPDS file No. 15-0755).
Furthermore, the thermal stability of the NiWO4 was evaluated by TGA/DTG, as shown in Fig. 1c. The weight loss (Δm1 = 16.96%) in the temperature range 61.7–257 °C is attributed to the desiccation of the moisture and gasses adsorbed on the surface. The desorption of water molecules is uninterrupted above 257 °C till near 604 °C with a small weight loss (Δm2 = 4.5%) because of the liberation of water molecules bonded in the structure of the sample and may be attributed to the decomposition of a small amount of grafted metal hydroxides. Beyond 640 °C, the nickel tungstate acquired heat stability and no weight loss was observed in the temperature area 604–1000 °C. DTG curves of the nickel tungstate showed a large and sharp endothermic peak in the temperature range (58–245 °C) corresponds to the desiccation of the sample. Further, small and wide endothermic peaks correlated with a small weight loss were obtained in the temperature range of 280–536 °C.
Eventually, morphological characterization was conducted using SEM, whereas size and shape of NiWO4 are depicted in Fig. 2a–c, where various images were pictured at different magnifications. The images revealed spherical morphology with relatively symmetrical dispersion. The EDX-mapping spectra are demonstrated in Fig. 2d–e to confirm the presence of constituent elements (nickel, tungsten, and oxygen) in the sample with an appreciable percentage. Figure 2f displayed the histogram of particle size distribution for chemically synthesized NiWO4.


SEM images (a–c); d EDX; e mapping and f corresponding particles size statistics results for NiWO4
4.2 Electrode modification and electrochemical measurements
Selection of a proper material for electrode modification is an important step to reach an high-performance voltammetric assay. Hence, different materials were fabricated, characterized and their voltammetric performances were tested. In this regard, the surface of the working electrode was covered with various types of metal/metal oxide composites. Mixed oxides of rare earth (Y, Sm, Ce) were calcined at 600 °C, the same mixed oxides were pyrolysed in the presence of N2 gas at 600 °C and the NiWO4 exhibited the highest contribution in the voltammetric peak current in comparison with the unmodified carbon paste electrode (Fig. 3A).

ACyclic voltammetric measurements showing the impact of modified electrodes on the redox reaction of FCN (10 mM). B FCN redox reactions at the superficies of the unmodified, MWCNs, NiWO4, and NiWO4/MWCNTs. C Generated voltammetric signals form the oxidation of deposited cadmium ions at the superficies of CPEs modified with MWCNTs, NiWO4, and NiWO4/MWCNTs. The Cd2+ level in the electrochemical cell was 10−4 M, scanning rate was 50 V/s.
Considerable attention was paid to the use of carbon nanotubes (CNTs) for the designing and optimizing electrochemical systems for having some advantages (high electrical conductivity, strong adsorptive ability and good mechanical strength [27]). Subsequently, cyclic voltammetry was performed using different working electrodes (either unmodified CPE, or modified CPEs with the MWCNTs, NiWO4, or NiWO4/MWCNTs composite).
As demonstrated in Fig. 3B, the incorporation of nanomaterial, especially multi-walled carbon nanotubes (MWCNTs) enhanced the electrode performance with improving the conductivity and accelerating the redox reactions taking place at the outer interface [28]. As a result, the composite of NiWO4/MWCNTs has evinced electro-catalytic activity and shifted the peak potential to the lower value as in Fig. 4. Furthermore, voltammetric signals of cadmium ions at the electrodes surface modified with the NiWO4/MWCNTs were much higher than that obtained by the unmodified or the CPE modified with the NiWO4, as illustrated in Fig. 3C. Accordingly, the use of the NiWO4/MWCNTs is very promising to be implemented in the direct voltammetric analysis of Cd ions.

Voltammetric responses of modified electrodes with various conecntrations of NiWO4. The given values are the ΔIoxid (oxidation peak current of cadmium deducted from the baseline of the electrolyte). The MWCNTs concentration was fixed at 2.0 mg/mL
4.3 Electrochemical sensing mechanism
At the start of the electrochemical reaction, Cd(II) ions are physically adsorbed on the modified electrode with the NiWO4.
Then, the target Cd(II) is reduced to Cd(0) under the applied DC-potential on the electrode surface
Eventually, the reduced Cd (0) is re-oxidized to Cd(II), and the oxidation peak is generated where the peak height is dependent on the cadmium concentration near to the modified electrode surface.
This method offers the detection of metal ions even at very low concentration with high sensitivity.
4.4 Optimizing the experimental conditions
Parameters that affect the electrochemical signals towards Cd(II) ions were optimized. Here, electrode composition, types of supporting electrolyte, pH, and scan rate are included. In the next subsections, results and discussion on each of these factors will be stated.
4.5 Effective concentration of modifier onto the electrode surface
To investigate the influence of different amounts of the sensing material (i.e. the NiWO4) on peak current of Cd(II), a series of concentrations of NiWO4 ranging from 0.0 to 8.0 mg/mL in presence of doping amount of multi-walled carbon nanotubes (2.0 mg/mL) was prepared and the voltammetric signals were investigated. As a result, Fig. 4 demonstrated that the oxidation peak current is strongly enhanced when the effective concentration of the materials was reached to 4.0 mg/mL. The increase in the oxidation current of cadmium ions was dependent on the increase of the modifier concentration on the modified electrode surface until reaching 6.0 mg/L. This voltammetric enhancement is attributed to the surface area expansions that supported the adsorption of Cd(II). However, further increase in the concentration of NiWO4 over 6.0 mg/mL caused a decrease in the oxidation peak current related to drop in the conductivity of NiWO4 modified CPE, and blocking in the electron transfer. Thus, 6.0 mg/mL of NiWO4 doped with 2.0 mg/mL of the multi-walled carbon nanotubes was chosen as an optimal concentration and has been applied to the next experiments.
On the other hand, estimation of active surface area of electrodes was performed using the Randles–Ševčík Eq. (1), as has been explained in Reference [5].
Where, IP is the density of oxidation current, n is the number of electrons transferred in a redox reaction of cadmium ions at the electrode surface, C is the cadmium concentration(s), A is the needed surface area to be identified (cm2), D is the diffusion coefficient of supporting electrolyte equals to 0.696*10−5 cm2S−1. Accordingly, the electrochemically active surface area of each of the carbon paste electrode was obtained as follows: 0.093, 0.203, 0.168, and 0.451 cm2 for the unmodified, MWCNTs, NiWO4, and NiWO4/MWCNTs modified electrodes, respectively. Besides, the generated current density was calculated for all electrode surfaces, and the results are reported as follows: 18, 41, 34, and 91µA for the unmodified, MWCNTs, NiWO4, and NiWO4/MWCNTs modified electrodes, respectively. Thus, the electrode modification with the selected NiWO4/MWCNTs enlarged the surface area and amplified the redox reaction taking place at the electrode surface.
4.6 Selection of supporting electrolyte
The influence of supporting electrolyte on the voltammetric determination of Cd(II) was studied in different prepared electrolytes including NaCl, phthalic acid, oxalic acid, phosphate buffer and acetate buffer. As shown in Fig. S1 (supplementary materials) phthalic acid has assisted the production of the highest anodic current. Therefore, phthalic acid having a pH value of 3.5 was selected as the optimum supporting electrolyte.
4.7 Effect of pH
The influence of pH on the peak current of Cd(II) was studied on the NiWO4/MWCNTs using Linear Sweep Voltammetry (LSV). As shown in Fig. 5, the peak current was increased with increasing the pH from 2.5 to 3.5 (within a very limited pH range). On the other hand, the anodic peak potential (Ep.a.) was shifted towards more negative potential with increasing the pH. A dramatic negative effect of pH on the peak current was observed when the pH value was above 3.5, where a sharp decrease was obtained indicating the sensitivity of the nanomaterials to the pH changes. Thus, for further study, supporting electrolyte with a pH 3.5 was chosen.

Influence of pH on the peak current and peak potential of cadmium ions (10−4 M) using the modified electrode with the selected material.
4.8 The influence of scan rate
To evaluate the electrochemical behavior of NiWO4/MWCNTs modified electrodes at different scanning rates, scan rate ranging from 30 to 300mV/s was tested at a fixed concentration of Cd(II). Figure 6 showed that the oxidation peak current is increased linearly with the increase of scanning rate.
Thus, peak currents were increased linearly as a function of the square root of the scan rate for reversible electron transfer. Plotting the obtained oxidation current (ip) vs. the square roots of scan rates (ν1/2) was applied for the characterization of electrochemically reversible redox systems according to the principle of Randles–Ševčík [12]. The height of peak current was directly dependent on the square root of scan rate, which reflects the homogeneous and fast electron transfer process with an adsorption-controlled electrochemical process. Moreover, altering of scan rates led to a shift in the oxidation peak potential to the positive direction to confirm the irreversibility of Cd(II) oxidation process at the modified CPEs [13, 29].
4.9 Calibration curve
Applying the optimum experimental conditions, a calibration curve was constructed by measuring the DPV responses for a series of standard solutions of Cd(II), in the concentration range 50–450 µM (as shown in Fig. 7. The peak current of Cd(II) displayed a linear relation with the regression coefficient (R2 = 0.989). The limit of detection (LOD), and limit of quantification (LOQ) for the Cd(II) were calculated from the calibration curves to be 0.12 µM, and 0.42µM, respectively. As shown in Table 1, some selected literatures are listed to show that the newly developed DPV assay using the modified CPE exhibited higher sensitivity compared with some other Cd2+ ion previously reported sensors.

(I) Influence of the scan rate on the alterations of peak currents. (II) Relationship between the square root of scan rate and the oxidation peak current of 10−4M of Cd(II). (III) Influence of the scan rate on the alterations of peak positions and peak currents

DPV peaks were registered using the chemically modified electrodes (I); Standard calibration curve for cadmium determination (II). All optimized parameters and conditions were applied
4.10 Effect of interferences
To assess the efficiency of the proposed sensing assay from the selectivity standpoint towards the electrochemical analysis of Cd(II)in the existence of other cations (e.g. Ag+, Mg2+, Zn2+, Cu2+, Co2+, and Fe3+). Individual experiments were carried out in 0.1 M phthalic acid (pH 3.5). Cd(II) was detected in the existence of aloft levels of the non-targeted metal ions mentioned above. There was no significant alteration either in the peak height or the peak for Cd(II) responses with the coexistence of high concentration of the studied metal ions, as shown in (Fig. 8). This assures that the fabricated sensor has good selectivity for the detection of Cd(II) and could be applied in a complex matrix environment.

(I) Anodic peak current of Cd(II) with concentration of 10−4M in presence high concentration of interferes ions. (II, III) Mg, Zn respectively interferes with 3-folds of the concentration of cadmium show no significant change in the peak potential and peak height.
4.11 Detection of cadmium in veritable samples
To evaluate the efficiency of the suggested voltammetric method, the proposed electrode was utilized to determine traces of the target element in the variety of collected real water sources (tap water, River-Nile water, ground water, and wastewater samples) and biological sample (urine sample). The standard addition method was performed under the optimum conditions for the determination of the unknown concentration of Cd(II) in samples. Determination of Cd(II) in a biological sample (urine sample) is performed after acid digestion. The obtained results have shown good recovery (90–108%) as illustrated in Table 2.
4.12 Conclusion
In this work, a carbon paste electrode modified with NiWO4-doped multi-walled carbon nanotubes nanocomposite was constructed for trace cadmium ion detection by differential pulse voltammetry (DPV). The NiWO4/MWCNTs composite revealed high sensitivity, better selectivity and strong adsorption ability for heavy metal cadmium ions during the deposition process. Sensing mechanisms were explored, effective parameters were identified, a wide dynamic linear range was obtained with high sensitivity, and ultra-high selectivity against several heavy metal ions. The obtained results exhibited a wide area of the concentration over the linear range of 50–450 µM with a limit of detection 0.12µM. The proposed electrode was successfully utilized for the estimation of levels of cadmium ions in real environmental and biological samples. Thus, the newly developed electrochemical method is in a continuous improvement process towards electrode modification and functionalization with nanomaterials.
References
Ding Q, Li C, Wang H, Xu C, Kuang H (2021) Chem Commun 57:7215
Shtenberg G, Massad-Ivanir N, Segal E (2015) Analyst 140:4507
Ismail A, Kawde A, Muraza O, Sanhoob MA, Al-Betar AR (2016) Microporous Mesoporous Mater 225:164
Mohammadi S, Taher MA, Beitollahi H, Naghizadeh M (2019) Environ Nanotechnol Monit Manag 12:100241
Rajab N, Ibrahim H, Hassan RYA, Youssef AFA (2023) RSC Adv 13(31):21259
Locatelli C, Torsi G (2003) Microchem J 75:233
Merlos Rodrigo MA, Cernei N, Kominkova M, Zitka O, Beklova M, Zehnalek J, Kizek R, Adam V (2013) Int J Environ Res Public Health 10:1304
Hassan RYA (2022) Sensors 22(19):7539
Magar HS, Hassan RYA, Mulchandani A (2021) Sensors 21:6578
Ismaiel AA, Aroua MK, Yusoff R (2014) Sensors 14:13102
Hezard T, Fajerwerg K, Evrard D, Collière V, Behra P, Gros P (2012) J Electroanal Chem 664:46
Magar HS, Magd EEAE, Hassan RYA, Fahim AM (2022) Microchem J 182:107885
Hassan RYA, Wollenberger U (2019) Electroanalysis 31:1112
Alfadaly RA, Elsayed A, Hassan RYA, Noureldeen A, Darwish H, Gebreil AS (2021) Molecules 26:2549
Sestakova I, Mader P (2000) Cell Mol Biol 46:257
Mekawy MM, Hassan RYA, Ramnani P, Yu X, Mulchandani A (2018) Arab J Chem 11(6):942–949
Afkhami A, Khoshsafar H, Bagheri H, Madrakian T (2014) Mater Sci Engineering: C 35:8
El-Fatah GA, Magar HS, Hassan RYA, Mahmoud R, Farghali AA, Hassouna MEM (2022) Sci Rep 12(1):20181
Moreno B, Chinarro E, Colomer MT, Jurado JR (2010) J Phys Chem C 114:4251
Wang W, Hu L, Ge J, Hu Z, Sun H, Sun H, Zhang H, Zhu H, Jiao S (2014) Chem Mater 26:3721
Nithya VD, Selvan RK, Kalpana D, Vasylechko L, Sanjeeviraja C (2013) ElectrochimicaActa 109:720
Kumar R, Bhuvana T, Sharma A (2017) RSC Adv 7:42146
Al-Othman ZA, Naushad M, Khan MR, Wabaidur SM (2012) J Inorg Organomet Polym Mater 22:352
AlShehri SM, Ahmed J, Alzahrani AM, Ahamad T (2017) New J Chem 41:8178
Pourmortazavi SM, Rahimi-Nasrabadi M, Khalilian-Shalamzari M, Zahedi MM, Hajimirsadeghi SS, Omrani I (2012) Appl Surf Sci 263 :745
Daemi S, Moalem-Banhangi M, Ghasemi S, Ashkarran AA (2019) J Electroanal Chem 848:113270
Yang N, Chen X, Ren T, Zhang P, Yang D (2015) Sens Actuators B 207:690
Anusha T, Bhavani KS, Kumar JVS, Brahman PK, Hassan RYA (2022) Bioelectrochemistry 143:107935
Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ (2012) Chem Rev 112:4016
Blaise N, Hambate Gomdje V, Maallah R, Oubaouz M, Tigana Djonse Justin B, Andrew Ofudje E, Chtaini A (2022) J Anal Methods Chem 2022:6900839. https://doi.org/10.1155/2022/6900839
Tamizhdurai P, Rajakumaran R, Sakthinathan S, Chen SM, Chiu T-W, Narayanan S (2020) Microporous Mesoporous Mater 307:110449
Bagheri H, Afkhami A, Khoshsafar H, Rezaei M, Shirzadmehr A (2013) Sens Actuators B 186:451
El Mhammedi MA, Achak M, Hbid M, Bakasse M, Hbid T, Chtaini A (2009) J Hazard Mater 170:590
Tamizhdurai P, Sakthinathan S, Krishnan PS, Ramesh A, Mangesh VL, Abilarasu A, Narayanan S, Shanthi K, Chiu T-W (2019) J Mol Struct 1176:650
Garg N, Deep A, Sharma AL (2023) Ind Eng Chem Res 62:9735
Yu L, Wan J-W, Meng X-Z, Gu H-W, Chen Y, Yi H-C (2022) Int J Environ Anal Chem 102:4757
Rahim AMA, Mahmoud EMM (2023) Anal Sci 39:179
Pu Y, Wu Y, Yu Z, Lu L, Wang X (2021) Talanta Open 3:100024
Hassan KM, Gaber SE, Altahan MF, Azzem MA (2020) Sens Bio-Sensing Res 29:100369
Zhang Y, Li C, Su Y, Mu W, Han X (2020) Inorg Chem Commun 111:107672
Lima TM, Soares PI, do Nascimento LA, Franco DL, Pereira AC, Ferreira LF (2021) Microchem J 168:106406
Antunović V, Ilić M, Baošić R, Jelić D, Lolić A (2019) PLoS ONE 14:e0210904
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
1—Prof. MEMH: conceptualization, writing—review & editing, visualization, project administration. 2—Prof. RYAH & Dr. RM: software, validation, formal analysis, resources, data curation, review the manuscript, supervision. 3—Prof. AAF: data curation, review the manuscript. 4—Mr. RHRM: methodology, software, formal analysis, investigation, resources, writing—original draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohammed, R.H.R., Hassan, R.Y.A., Mahmoud, R. et al. Electrochemical determination of cadmium ions in biological and environmental samples using a newly developed sensing platform made of nickel tungstate-doped multi-walled carbon nanotubes. J Appl Electrochem 54, 657–668 (2024). https://doi.org/10.1007/s10800-023-01976-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-023-01976-y