
Abstract
Brahma-related gene 1 (BRG1) has been implicated in the repair of DNA double-strand breaks (DSBs). Downregulation of BRG1 impairs DSBs repair leading to accumulation of double-stranded DNA (dsDNA). Currently, the role of BRG1 in diabetic cardiomyopathy (DCM) has not been clarified. In this study, we aimed to explore the function and molecular by which BRG1 regulates DCM using mice and cell models. We found that BRG1 was downregulated in the cardiac tissues of DCM mice and in cardiomyocytes cultured with high glucose and palmitic acid (HG/PA), which was accompanied by accumulation of dsDNA and activation of the cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) signaling pathway. shRNA-mediated Brg1 knockdown aggravated DCM mice cardiac functions, enhanced dsDNA accumulation, cGAS-STING signaling activation, which induced inflammation and apoptosis. In addition, the results were further verified in HG/PA-treated primary neonatal rat cardiomyocytes (NRCMs). Overexpression of BRG1 in NRCMs yielded opposite results. Furthermore, a selective cGAS inhibitor RU.521 or STING inhibitor C-176 partially reversed the BRG1 knockdown-induced inflammation and apoptosis in vitro. In conclusion, our results demonstrate that BRG1 is downregulated during DCM in vivo and in vitro, resulting in cardiomyocyte inflammation and apoptosis due to dsDNA accumulation and cGAS-STING signaling activation. Therefore, targeting the BRG1-cGAS-STING pathway may represent a novel therapeutic strategy for improving cardiac function of patients with DCM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes, a non-communicable metabolic disease, is increasingly becoming a major pandemic worldwide [1]. Although great progress has been made in the establishment of diabetes treatment, the available treatments have several chronic complications, especially cardiovascular diseases [2]. DCM is the main cause of death in diabetic patients, and is characterized by myocardial hypertrophy, cardiac fibrosis, and heart failure [3]. The pathophysiology of DCM is complex with multiple pathophysiological mechanisms associated with its, including mitochondrial dysfunction, myocardial inflammation, and apoptosis [4, 5]. Uncontrolled myocardial inflammation and apoptosis are key processes that cause cardiac dysfunction in DCM [6]. Therefore, expanding our understanding of the complexity of DCM pathophysiology, particularly the identification of novel genes and regulatory pathways, will help to identify treatments for DCM-induced cardiomyocyte inflammation and apoptosis.
BRG1, also known as SMARCA4, encodes a component of the switch/sucrose nonfermentable (SWI/SNF) complex, which regulates the ATPase and helicase activities. Recent studies have demonstrated that BRG1 downregulation increased the transcription of proinflammatory genes and induced apoptosis [7, 8]. A previous study demonstrated that BRG1 participates in cardiac growth and differentiation processes [9]. In our earlier study, we found that BRG1 was upregulated during acute myocardial infarction, and BRG1 overexpression alleviated cardiomyocyte oxidative damage and increased cardiomyocyte viability [10]. In addition, BRG1 upregulation was found to ameliorate diabetic cardiomyopathy-induced diastolic dysfunction [11]. However, whether BRG1 is involved in the development of DCM remains to be determined.
Studies have implicated BRG1 in the repair of DNA double-strand breaks (DSBs), loss of BRG1 impairs DSBs repair resulting in the accumulation of cytoplasmic dsDNA [12, 13]. cGAS, a dsDNA sensor, uses cytosolic DNA to generate cyclic GMP–AMP, which binds to its receptor STING. Subsequently, STING activates the transcription factor nuclear factor-kappa B (NF-κB) and interferon regulatory factor 3 (IRF3) via the TANK binding kinase 1 (TBK1) [14, 15]. Recent studies have confirmed that cGAS–STING also contributes to DCM by sensing mitochondrial damage-released DNA [16, 17]. Therefore, studies are needed to explore the relationship between BRG1 expression and the cGAS–STING during the pathogenesis of DCM.
In this study, we utilize an HFD and streptozotocin (STZ)-induced DCM mouse model and HG/PA-treated cardiomyocytes injury model to investigate the relationship between BRG1 expression and cGAS-STING activation in DCM. BRG1 was knocked down in DCM mice and HG/PA-treated cardiomyocytes to deeply explore the mechanism by which BRG1 regulates DCM. These results show that BRG1 has promising therapeutic potential for application in DCM.
Materials and Methods
Vector and Adeno-associated Virus; Lentivirus Construction and Adenovirus
The adeno-associated virus (AAV) vector carrying cardiac troponin T (CTNT) promoter, green fluorescent protein (GFP) and Brg1 shRNA were purchased from Hanbio Biotechnology Co. Ltd (Shanghai, China). The cardiomyocyte-specific CTNT promoter promotes the expression of Brg1 shRNA in cardiomyocytes. The lentiviral virus vector carrying GFP and Brg1 shRNA was constructed by Cyagen Biosciences (Guangzhou, China). The Brg1 shRNA sequence was 5′-GCTGCCAAATACAAACTCAATCTCGAGATTGAGTTTGTATTTGGCAGC-3′. The adenovirus carrying GFP and Brg1-adenovirus were constructed by the WZ Biosciences Inc. (Shandong, China). The titers of the AAV stock, lentivirus stock and adenovirus stock were 1.68 × 1012 vg/ml, 3.98 × 109 TU/ml and 1 × 1010 pfu/ml, respectively.
Animal Experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee of Affiliated Qingyuan Hospital, Guangzhou Medical University (Guangdong, China). 3–4-weeks old male C57BL/6J mice were purchased from Vital River Laboratory Animal Technology Co., Ltd (Guangdong) and housed under specific pathogen-free condition with a 12 h light/dark cycle, and 25 ± 1 °C, 60 ± 5% humidity. The mice were randomly divided into six groups: control (CON), control with AAV 9-scramble (AAV-scramble), control with myocardium-specific knockdown of Brg1 shRNA (AAV-Brg1 shRNA), DCM, DCM with AAV-scramble, and DCM with AAV-Brg1 shRNA. They were then intravenously injected with AAV-Brg1 shRNA into the tail vein to establish a model of myocardium-specific knockdown of Brg1. The diabetic mice were subsequently intraperitoneally injected with streptozotocin (STZ; 85 mg/kg, Sigma Aldrich, USA) twice and fed on HFD. Mice in the control group were given a normal diet and injected with the same volume of citrate sodium buffer. After injection, a contour glucose meter (Johnson & Johnson, USA) was used to measure blood glucose levels at 3, 5, and 7 days, and postprandial blood glucose ≥ 16.7 mmol/L indicated diabetes. After 16 weeks of treatment, cardiac function was evaluated using the Vevo2100 system.
Echocardiography
Mice electrocardiography was performed using a transthoracic echocardiography (Vevo2100; Visual Sonics, Canada). Briefly, mice were anesthetized with 2% isoflurane, and cardiac function parameters were measured, including the E/A ratio, left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS), and other left ventricular (LV) parameters.
Isolation of NRCMs and Treatment
The NRCMs were isolated from 1–2 days Sprague Dawley rats as previously described [18]. Cultured cardiomyocytes were transfected with Ad-Mock, Ad-Brg1 WT [the multiplicity of infection (MOI) = 5]; Lenti-scramble, or Lenti-Brg1 shRNA (the MOI = 25) for 48 h, or pretreated with 10 µmol RU.521 (Cat. #HY-114180; New Jersey city, USA) or C-176 (Cat. # HY-112906; New Jersey city, USA) for 24 h and then incubated with 33 mM glucose and 300 μM palmitic acid for 48 h. The NRCMs were subsequently harvested for analysis.
Histology and Immunofluorescent Staining
Fresh mice hearts were fixed in 10% formalin solution and embedded in paraffin. The heart was sectioned into 4 μm thick sections and stained with Masson's and hematoxylin-eosin (HE) staining.
In vitro, cardiomyocytes were fixed in 4% paraformaldehyde. The membranes were permeabilized with 1% triton and incubated with 100 nM glycine. Subsequently, they were blocked with 10% goat serum for 1 h followed by incubation with primary antibody overnight at 4 °C. On the following day, cardiomyocytes or tissue sections were incubated with dylight 561-coupled anti-mouse IgG and dylight 488/647-coupled anti-rabbit IgG. Finally, the nuclei were stained with DAPI at a concentration of 0.5 μg/ml. The sections were imaged using a confocal microscope (LSM900). The antibody information is presented in Table S1.
Western Blotting (WB) Assessment
Cells or tissues were lysed to extract proteins, which was then separated by 10–15% sodium dodecyl-sulfate polyacrylamide gel electrophoresis gel (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane. The PVDF membranes were blocked with 5% skimmed milk and incubated with primary antibodies overnight at 4 °C. The membranes were then incubated with a secondary antibody. Finally, the protein bands were analyzed using the gel imaging system (Biorad ChemiDoc). The details of antibodies used are presented in Table S1.
Quantitative Real-time PCR (qRT-PCR)
The total RNA was extracted from treated cardiomyocytes or cardiac tissues using TRIzol reagent (Invitrogen). The RNA was reverse transcribed into complementary DNA (cDNA) using the Takara PrimeScript™ RT Master Mix (Takara Bio Inc., Japan). The cDNA was subjected to qPCR amplification on the CFX Connect™ Real-Time system (Biorad). The primers used in this experiment were designed by the NCBI Primer-BLAST and are listed in Table S2.
TdT-mediated dUTP Nick End Labeling (TUNEL) Staining
The apoptosis rate of cardiomyocytes was determined by TUNEL staining. Briefly, mice myocardial tissues were excised, fixed, paraffinized and then sectioned. In vitro, cardiomyocytes were fixed by 4% paraformaldehyde solution. The membranes were permeabilized with 1% triton and incubated with 100 nM glycine. The incubation buffer was the prepared following instructions on the TUNEL kit (Cat. 12156792910 Roche Diagnostics, Germany), and was applied in a constant temperature incubator at 37 °C in darkness. The nuclei were stained with DAPI at a concentration of 0.5 μg/ml. Confocal microscope (LSM900) was employed to examine and capture images of the slices for analysis.
Statistical Analysis
Statistical analysis was performed by GraphPad Prism 8.0 (San Diego, CA, USA), and the data were expressed as the mean ± standard error. Groups were compared with paired t-tests or one-way ANOVAs with post-hoc Tukey tests. P < 0.05 was considered statistically significant.
Results
BRG1 is Downregulated in the Heart of DCM Mice
In this study, we established a DCM mouse model by feeding a HFD and administering STZ. We initially assessed the BRG1 protein levels in the cardiac tissues of DCM mice. Western blot analysis revealed that BRG1 protein level was decreased in the cardiac tissues of DCM mice compared to normal mice (Fig. 1a). Immunofluorescence staining further confirmed the localization and decreased expression of BRG1 in the hearts of DCM mice (Fig. 1b). Considering that BRG1 participates in the repair of DSBs, we detected the accumulation of H2AX phosphorylation (γ-H2AX, a surrogate marker for DSBs) and dsDNA in the mice cardiac tissues. It was observed that the γ-H2AX protein level was higher in cardiac tissues from the DCM mice compared with the normal mice (Fig. 1a). In addition, the cytoplasmic dsDNA content was increased in the heart tissues (Fig. 1c). Consistent with these results, the protein levels of cGAS, STING, p-TBK, and p-NF-κB were significantly upregulated in cardiac tissues from the DCM mice (Fig. 1d). Furthermore, analysis of the expression level of inflammation and apoptosis markers in the pathogenesis of DCM revealed that the concentration of IL-1β and cleaved caspase-3 were upregulated in the cardiac tissues from DCM mice (Fig. 1e and f). These results suggested that BRG1 was downregulated in the heart tissues of DCM model, which may result in cGAS-STING signaling activation, inflammation and apoptosis.

BRG1 is downregulated in the heart of DCM mice. a Protein levels of BRG1 and γ-H2AX were assayed using Western blot in mice cardiac tissues. b Representative immunofluorescence images of BRG1 in cardiac tissues. c The accumulation of dsDNA was evaluated using immunofluorescent staining in mice cardiac tissues. d Protein levels of cGAS-STING signaling-related genes were assayed using Western blot in mice cardiac tissues. e Level of IL-1β protein was assayed using Western blot in mice cardiac tissues. f Level of cleaved caspase-3 was assayed using Western blot in mice cardiac tissues. * indicates P < 0.05 vs. the CON group.
BRG1 Expression is Downregulated in HG/PA-treated Cardiomyocytes
Primary cardiomyocytes were incubated with HG/PA to mimic the hyperglycemia and hyperlipemia in vitro. Subsequently, the expression levels of BRG1 and γ-H2AX in the HG/PA-treated cardiomyocytes were measured. Compared with the control group, BRG1 expression was gradually decreased in a time-dependent manner following HG/PA stimulation, whereas the expression of γ-H2AX was gradually increased (Fig. 2a). The immunofluorescence results confirmed the localization and downregulation of BRG1 expression in HG/PA-cultured cardiomyocytes (Fig. 2b). Furthermore, we evaluated the cytoplasmic dsDNA content and the activity of the cGAS–STING in the HG/PA-treated cardiomyocytes. Data shown in Fig. 2c suggested that the cytoplasmic dsDNA content was increased following HG/PA treatment. In addition, the protein levels of cGAS, STING, p-TBK, and p-NF-κB were significantly upregulated in HG/PA-treated cardiomyocytes (Fig. 2d). Consistent with this, the expression levels of IL-1β and cleaved caspase-3 were upregulated in HG/PA-treated cardiomyocytes in a time-dependent manner (Fig. 2e and f). Altogether, these results indicated that BRG1, dsDNA, and the cGAS–STING co-regulate inflammation and apoptosis in HG/PA-treated cardiomyocytes.

BRG1 expression is downregulated in HG/PA-treated cardiomyocytes. a Protein levels of BRG1 and γ-H2AX were assayed using Western blot. b Representative immunofluorescence images of BRG1 in cardiomyocytes. c The accumulation of dsDNA was evaluated using fluorescent staining. d Protein levels of cGAS-STING signaling-related genes were assayed using Western blot. e Level of IL-1β protein was assayed using Western blot. f Level of cleaved caspase-3 protein was assayed using Western blot. * indicates P < 0.05 vs. the CON group.
BRG1 Deficiency Aggravated Cardiac Dysfunction In Vivo
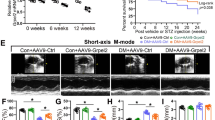
To explore the function of BRG1 in DCM pathogenesis, we constructed a DCM mouse model with myocardium-specific knockdown of BRG1 via injection of AAV-Brg1 shRNA. The experimental design of this process is shown in Fig. 3a. The GFP-tagged virus was used as a scrambled control. The in vivo imaging and GFP immunofluorescence results indicated that AVV targeted and was enriched in myocardial tissue (Supplementary Fig. 1A and B). In addition, the WB results confirmed that the protein level of BRG1 in heart tissue was effectively knocked down following AAV-Brg1 shRNA injection (Fig. 3b). The heart weight/tibial length (HW/TL) ratio (Fig. 3c) and the sectional area of myocardial cross (Fig. 3d) were increased after AAV-Brg1 shRNA injection. Analysis of the echocardiography results revealed significant reduction in the E/A ratio in the model group. Mice injected with AAV-Brg1 shRNA exhibited significantly decreased in the E/A ratio compared to the control group (Fig. 3g).Compared to the DCM group, mice injected with AAV-Brg1 shRNA exhibited significantly decreased echocardiographic parameters of LVEF, LVFS, E/A ratio, and LVPW in end-systole, while LVID in end-systole was significantly increased (Fig. 3e-i). In addition, the heart rates were comparable among the study groups (Fig. 3j). These results indicated that BRG1 contributed to the pathological process of DCM in vivo.

BRG1 deficiency aggravates experimental mouse cardiac dysfunction in vivo. a Schematic illustration for mouse experimental design. b Level of BRG1 protein was assayed using Western blot in mice cardiac tissues. c Calculation and analysis of the heart weight/tibial length (HW/TL) ratio. d The HE staining and Masson's staining of the mouse heart tissues. e Representative M-mode echocardiographic imaging. f Calculation of ejection fraction and fractional shortening of heart. g E/A ratio. h left ventricular posterior wall (LVPW) in end-systole. i left ventricular internal diameter (LVID) in end-systole. j Heart rate. * indicates P < 0.05 vs. the CON group. # indicates P < 0.05 vs. the DCM group.
BRG1 Deficiency Increased Cytoplasmic dsDNA Accumulation and Activated cGAS-STING Signaling
To explore the role of BRG1 in the pathological mechanism of DCM, we assessed γ-H2AX expression, cytoplasmic dsDNA content, and activation of the cGAS-STING in DCM mice following cardiomyocyte-specific BRG1 knockdown. Our results revealed a significant increase in the protein level of γ-H2AX following AAV-Brg1 shRNA injection (Fig. 4a). Consistent with this finding, the cytoplasmic dsDNA content was also increased after AAV-Brg1 shRNA transfection (Fig. 4b). Furthermore, AAV-Brg1 shRNA significantly upregulated the protein levels of cGAS, STING, p-TBK, and p-NF-κB in both control and DCM mouse cardiac tissues (Fig. 4c). Collectively, these results suggested that BRG1 knockdown increased the cytoplasmic dsDNA content and activated the cGAS-STING.

BRG1 deficiency increased cytoplasmic dsDNA accumulation and activated cGAS-STING signaling in mice cardiac tissues. a Level of γ-H2AX protein was assayed using Western blot in mice cardiac tissues. b The accumulation of dsDNA was evaluated using fluorescent staining. c Protein levels of cGAS-STING signaling-related genes were assayed using Western blot in mice cardiac tissues. * indicates P < 0.05 vs. the CON group, # indicates P < 0.05 vs. the DCM group.
BRG1 Deficiency Induced Inflammation and Apoptosis in Mouse Cardiac Tissues
Since dsDNA and cGAS-STING have close relationship with inflammation and apoptosis, we detected the concentration of markers in the cardiac tissues of AAV-Brg1 shRNA transfected mice. The WB results indicated that the IL-1β protein level was significantly upregulated in the AAV-Brg1 shRNA transfected group (Fig. 5a). Levels of tnf-α and il-6 mRNA also exhibited similar trends (Fig. 5b). TUNEL straining results confirmed that knockdown of BRG1 increased the percentage of apoptotic cardiomyocytes (Fig. 5c). In addition, cleaved caspase-3 was upregulated in cardiac tissues of AAV-Brg1 shRNA transfected mice (Fig. 5d). These findings demonstrated that knockdown of BRG1 induced inflammation and apoptosis in mice cardiac tissues.

BRG1 deficiency induced inflammation and apoptosis in mouse cardiac tissues. a Level of IL-1β was assayed using Western blot in mice cardiac tissues. b mRNA levels of tnf-α and il-6 were evaluated using qRT-PCR. c The apoptosis was detected by the TUNEL assay in mice cardiac tissues. d Level of cleaved caspase-3 was assayed using Western blot. * indicates P < 0.05 vs. the CON group, # indicates P < 0.05 vs. the DCM group.
BRG1 Overexpression Inhibited HG/PA-induced Cytoplasmic dsDNA Accumulation and cGAS-STING Signaling Activation I n Vitro
Further, we employed NRCMs to verify the in vivo results. The WB results indicated that HG/PA treatment downregulated the protein level of BRG1 and upregulated the protein level of γ-H2AX in cardiomyocytes, which was consistent with the in vivo results (Fig. 6a and b). Moreover, HG/PA treatment increased the cytoplasmic dsDNA content in cardiomyocytes (Fig. 6c and d). We also observed that the HG/PA treatment significantly upregulated the protein levels of cGAS, STING, p-TBK and p-NF-κB in cardiomyocytes (Fig. 6e and f).

BRG1 overexpression inhibited HG/PA-induced cytoplasmic dsDNA accumulation and cGAS-STING signaling activation in vitro. a, b Protein levels of BRG1 and γ-H2AX in cardiomyocytes were assayed using Western blot. c, d The accumulation of dsDNA was evaluated using fluorescent staining. e, f Protein levels of cGAS-STING signaling-related genes were assayed using Western blot. * indicates P < 0.05 vs. the CON group, # indicates P < 0.05 vs. the HG/PA group.
To assess the impact of BRG1 on cytoplasmic dsDNA content and activation of the cGAS-STING, we manipulated BRG1 expression in cardiomyocytes through adenovirus or lentivirus transfection for upregulation or downregulation, respectively. Knockdown of BRG1 further increased the HG/PA-induced upregulation of γ-H2AX, while BRG1 overexpression had the opposite effect (Fig. 6a and b). In addition, BRG1 knockdown enhanced HG/PA-induced cytoplasmic dsDNA accumulation, while BRG1 overexpression showed an opposite effect (Fig. 6c and d). Consistent with this, BRG1 knockdown increased the protein levels of cGAS, STING, p-TBK and p-NF-κB, whereas BRG1 overexpression had an opposite effect (Fig. 6e and f). These findings strongly suggest that BRG1 deficiency results in cytoplasmic dsDNA accumulation and cGAS-STING signaling activation.
BRG1 Overexpression Reduction of Inflammation and Apoptosis In Vitro
Next, we evaluated the function of BRG1 in HG/PA-induced cardiomyocyte inflammation and apoptosis. The WB results indicated that BRG1 knockdown further enhanced the IL-1β protein expression in HG/PA-managed cardiomyocyte, whereas BRG1 overexpressing had an opposite effect (Fig. 7a and c). The levels of tnf-α and il-6 mRNA also exhibited similar trends (Fig. 7b and d). In addition, the TUNEL staining results showed that BRG1 deficiency further enhanced the apoptosis of cardiomyocytes, while BRG1 overexpression showed the opposite effect (Fig. 7e). Similarly, BRG1 knockdown upregulated the expression of cleaved caspase-3 protein, while BRG1 overexpression triggered opposite effect (Fig. 7f). These results indicated that BRG1 overexpression inhibited HG/PA-induced cardiomyocyte inflammation and apoptosis.

BRG1 overexpression reduction of inflammation and apoptosis in vitro. a, c Protein level of IL-1β was assayed using Western blot in cardiomyocytes. b, d mRNA levels of tnf-α and il-6 were evaluated using qRT-PCR. e The apoptosis of cardiomyocytes was detected by the TUNEL assay. f Protein level of cleaved caspase-3 was assayed using Western blot. * indicates P < 0.05 vs. the CON group, # indicates P < 0.05 vs. the HG/PA group.
Blockade of cGAS-STING Attenuated BRG1 Downregulation-induced Cardiomyocyte Inflammation and Apoptosis
To further elucidate the role of cGAS-STING in BRG1 downregulation-induced cardiomyocyte injury in vitro, the activity of cGAS-STING was inhibited using selective inhibitors in BRG1 knockdown and HG/PA-treated cardiomyocytes. WB analysis showed that RU.521 or C-176, a selective inhibitor for cGAS or STING, successfully inhibited cGAS or STING expression (Supplementary Fig. 2A and B).
Data presented in Fig. 8a indicates that both RU.521 and C-176 blocked BRG1 knockdown-induced upregulation of IL-1β and cleaved caspase-3, while RU.521 or C-176 had no effect on the expression of BRG1 and γ-H2AX (Fig. 8a). Moreover, RU.521 and C-176 inhibition alleviated apoptosis in Lenti-Brg1 shRNA-infected NRCMs (Fig. 8b). These results suggested that BRG1 knockdown enhanced the HG/PA-induced cardiomyocyte inflammation and apoptosis via the cGAS-STING pathway.

BRG1 regulates cardiomyocyte inflammation and apoptosis through cGAS-STING signaling. a Protein levels of BRG1, γ-H2AX, IL-1β and cleaved caspase-3 were assayed using Western blot in cardiomyocytes. b The apoptosis of cardiomyocytes was detected by the TUNEL assay. c Illustration of the proposed signaling under activation of the BRG1 deficiency mediated cGAS-STING pathway in DCM. * indicates P < 0.05 vs. the CON group, # indicates P < 0.05 vs. the HG/PA group, † indicates P < 0.05 vs. the HG/PA + Lenti-Brg1 shRNA group.
Based on these results, we speculate that a hyperglycemic and hyperlipemic internal environment downregulates BRG1. This BRG1 deficiency activates the cGAS-STING by inducing dsDNA accumulation, thereby leading to cardiomyocyte inflammation and apoptosis (Fig. 8c).
Discussion
DCM is characterized by increased cardiomyocyte apoptosis and hypertrophy, as accompanied by impaired cardiac function, which elevates the risk of HF and sudden death in DCM patients [19]. In some preliminary studies, treatments such as sodium-glucose cotransporter 2 inhibitors and dipeptidyl peptidase-4 inhibitors were found to have the potential to treat DCM [20,21,22]. However, their clinical efficacy is limited. Therefore, there is an urgent need to explore the mechanisms of DCM and develop effective interventions to improve the DCM symptoms. In this study, we found that BRG1 deficiency led to dsDNA accumulation, and activation of the cGAS-STING signaling in cardiomyocytes from DCM mouse model and HG/PA-cultured NRCMs. BRG1 downregulation aggravated cardiomyocyte dysfunction, resulting in cardiomyocyte inflammation and apoptosis by enhancing dsDNA accumulation and activating cGAS-STING signaling (see Fig. 8c).
BRG1 has been implicated in the pathogenesis of cardiovascular disease [23]. A previous study found that BRG1 attenuated exercise-induced physiological myocardial hypertrophy by inhibiting pressure overload-induced histone deacetylase 2 activation and serine/threonine kinase/glycogen synthase kinase 3β phosphorylation [24]. A study by Funamoto et al. reported that the p300/BRG1 complex promotes occurrence of heart failure by enhancing the histone globular domain H3K122 acetylation [25]. In cultured endothelial cells and arteries, proinflammatory stimuli augmented the expression of BRG1. Previous investigations have demonstrated that BRG1 contribute to inflammation and endothelial NO synthase phosphorylation thereby contributing to the development of atherosclerosis [26, 27]. Moreover, BRG1 suppressed neutrophil infiltration and modulated NO bioavailability in endothelial cells to inhibit cardiac ischemia–reperfusion injury in mice [28, 29]. In our previous study, we found that BRG1 protected the heart against acute myocardial infarction and reduced oxidative damage by activating the NRF2/HO1 signaling pathway [10]. In addition, upregulation of BRG1 expression ameliorated hyperglycemia-induced oxidative stress and cardiac hypertrophy [11, 30]. In this study, we found that BRG1 deficiency promoted the progression of DCM and aggravated cardiac dysfunction in vivo, demonstrating that BRG1 is a potential therapeutic target of DCM.
The pathogenesis of DCM is multifaceted, with inflammation and apoptosis emerging as significant factors. Given the limited capacity for cardiomyocyte proliferation in the adult human heart, apoptosis of cardiac muscle cells stands out as a primary contributor to cardiac remodeling and dysfunction [31, 32]. BRG1, a protein involved in various biological processes including apoptosis and inflammation, is also implicated in DCM. A recent study indicated that BRG1 overexpression attenuates apoptosis induced by high glucose exposure in retinal ganglion cells through Notch activation [33]. In addition, increased BRG1 levels have been associated with reduced inflammatory responses and decreased oxidative damage in cerebral ischemia–reperfusion injury, and BRG1 deficiency has been linked to inflammation-driven colorectal cancer [8, 34]. As a core subunit of the SWI/SNF complex, BRG1 promotes the DSBs repair by stimulating the γ-H2AX at the DSB-surrounding chromatin. Loss of BRG1 promotes DSBs repair and upregulates γ-H2AX [12, 35]. Moreover, we found that BRG1 deficiency resulted in upregulation of γ-H2AX expression in vivo and in vitro.
cGAS-STING is an evolutionarily conserved defense mechanism hat senses pathogenic DNA, and triggers the innate immune reaction by stimulating type I interferon secretion [36]. In addition, the cGAS-STING was reported to be involved in dsDNA-induced inflammation and apoptosis [37, 38]. In a previous study, doxorubicin increased the DNA damage in cardiac endothelial cells causing accumulation of dsDNA fragments and activation of cGAS-STING pathway [39]. Another recent study found that γ-H2AX upregulation may indirectly increase dsDNA accumulation in the cytoplasm [13]. In our study, we confirmed that BRG1 downregulation increased γ-H2AX expression, accompanied by the dsDNA accumulation and activation of the cGAS-STING to induce cardiomyocyte inflammation and apoptosis. A previous study indicated that failure to repair and eliminate DSBs promptly leads to the accumulation of dsDNA and subsequent apoptosis. Inhibition of cGAS has been shown to reduce cardiomyocyte apoptosis [40]. Additionally, another study demonstrated that myocardial infarction results in the release of cardiac dsDNA, and inhibiting the STING can alleviate cardiomyocyte apoptosis [41]. Our results further showed that both cGAS and STING inhibition alleviated the BRG1 knockdown-induced upregulation of IL-1β and cleaved caspase-3 in NRCMs, thereby preventing cardiomyocyte apoptosis. Together, these data demonstrate that BRG1 deficiency modulates cardiomyocyte inflammation and apoptosis by activating the cGAS-STING pathway.
Collectively, our results demonstrated that BRG1 is downregulated in hyperglycemic and hyperlipemic cardiomyocytes both in vivo and in vitro. We found that BRG1 deficiency resulted in the accumulation of dsDNA and triggered cGAS-STING activation, exacerbating cardiomyocyte inflammation and apoptosis induced by hyperglycemia and hyperlipemia. These findings suggest a potential novel therapeutic approach for managing cardiomyocyte injury in DCM.
Data Availability
No datasets were generated or analysed during the current study.
References
Sun, H., P. Saeedi, S. Karuranga, M. Pinkepank, K. Ogurtsova, B.B. Duncan, C. Stein, A. Basit, J.C.N. Chan, J.C. Mbanya, et al. 2022. IDF Diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Research and Clinical Practice 183: 109119.
Greenlee, M.C., S. Bolen, W. Chong, A. Dokun, J. Gonzalvo, M. Hawkins, W.H. Herman, E. Leake, B. Linder, and P.R. Conlin. 2023. The national clinical care commission report to congress: leveraging federal policies and programs to improve diabetes treatment and reduce complications. Diabetes Care 46 (2): 51–59.
Pop-Busui, R., J.L. Januzzi, D. Bruemmer, S. Butalia, J.B. Green, W.B. Horton, C. Knight, M. Levi, N. Rasouli, and C.R. Richardson. 2022. Heart failure: an underappreciated complication of diabetes. A consensus report of the american diabetes association. Diabetes Care 45 (7): 1670–1690.
Tan, Y., Z. Zhang, C. Zheng, K.A. Wintergerst, B.B. Keller, and L. Cai. 2020. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nature Reviews Cardiology 17 (9): 585–607.
Ritchie, R.H., and E.D. Abel. 2020. Basic mechanisms of diabetic heart disease. Circulation Research 126 (11): 1501–1525.
Ramesh, P., J.L. Yeo, E.M. Brady, and G.P. McCann. 2022. Role of inflammation in diabetic cardiomyopathy. Therapeutic Advances in Endocrinology and Metabolism 13: 20420188221083530.
Mao, X., B. Yan, H. Chen, P. Lai, and J. Ma. 2023. BRG1 mediates protective ability of spermidine to ameliorate osteoarthritic cartilage by Nrf2/KEAP1 and STAT3 signaling pathway. International Immunopharmacology 122: 110593.
Guo, K., Y. Shang, Z. Wang, Y. Li, J. Chen, B. Zhu, D. Zhang, and J. Chen. 2012. BRG1 alleviates microglial activation by promoting the KEAP1-NRF2/HO-1 signaling pathway and minimizing oxidative damage in cerebral ischemia–reperfusion. International Immunopharmacology 119: 110201.
Hang, C.T., J. Yang, P. Han, H.-L. Cheng, C. Shang, E. Ashley, B. Zhou, and C.-P. Chang. 2010. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 466 (7302): 62–67.
Liu, X., X. Yuan, G. Liang, S. Zhang, G. Zhang, Y. Qin, Q. Zhu, Q. Xiao, and N. Hou. 2020. Luo J-d: BRG1 protects the heart from acute myocardial infarction by reducing oxidative damage through the activation of the NRF2/HO1 signaling pathway. Free Radical Biology and Medicine 160: 820–836.
Li, H., W. Yao, M.G. Irwin, T. Wang, S. Wang, L. Zhang, and Z. Xia. 2015. Adiponectin ameliorates hyperglycemia-induced cardiac hypertrophy and dysfunction by concomitantly activating Nrf2 and Brg1. Free Radical Biology and Medicine 84: 311–321.
Qi, W., R. Wang, H. Chen, X. Wang, T. Xiao, I. Boldogh, X. Ba, L. Han, and X. Zeng. 2015. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. Journal of Cell Science 128 (2): 317–330.
Han, X., H. Chen, H. Gong, X. Tang, N. Huang, W. Xu, H. Tai, G. Zhang, T. Zhao, C. Gong, et al. 2020. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress–induced senescence. Journal of Biological Chemistry 295 (14): 4451–4463.
Decout, A., J.D. Katz, S. Venkatraman, and A. Ablasser. 2021. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nature Reviews Immunology 21 (9): 548–569.
Skopelja-Gardner, S., J. An, and K.B. Elkon. 2022. Role of the cGAS–STING pathway in systemic and organ-specific diseases. Nature Reviews Nephrology 18 (9): 558–572.
Ma, X.M., and K. Geng. 2022. Law BY-K, Wang P, Pu YL, Chen Q, Xu HW, Tan XZ, Jiang ZZ, Xu Y: Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biology and Toxicology 39 (1): 277–299.
Yan, M., Y. Li, Q. Luo, W. Zeng, X. Shao, L. Li, Q. Wang, D. Wang, Y. Zhang, H. Diao, et al. 2022. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS–STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discovery 8 (1): 258.
Hou, N., B. Cai, C.-W. Ou, Z.-H. Zhang, X.-W. Liu, M. Yuan, G.-J. Zhao, S.-M. Liu, L.-G. Xiong, J.-D. Luo, et al. 2017. Puerarin-7-O-glucuronide, a water-soluble puerarin metabolite, prevents angiotensin II-induced cardiomyocyte hypertrophy by reducing oxidative stress. Naunyn-Schmiedeberg’s Archives of Pharmacology 390 (5): 535–545.
Dhar, A., J. Venkadakrishnan, U. Roy, S. Vedam, N. Lalwani, K.S. Ramos, T.K. Pandita, and A. Bhat. 2023. A comprehensive review of the novel therapeutic targets for the treatment of diabetic cardiomyopathy. Therapeutic Advances in Cardiovascular Disease 17: 17539447231210170.
Paolillo, S., F. Marsico, M. Prastaro, F. Renga, L. Esposito, F. De Martino, P. Di Napoli, I. Esposito, A. Ambrosio, M. Ianniruberto, et al. 2019. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Failure Clinics 15 (3): 341–347.
Zhang, L., H. Zhang, X. Xie, R. Tie, X. Shang, Q. Zhao, J. Xu, L. Jin, J. Zhang, and P. Ye. 2023. Empagliflozin ameliorates diabetic cardiomyopathy via regulated branched-chain amino acid metabolism and mTOR/p-ULK1 signaling pathway-mediated autophagy. Diabetology and Metabolic Syndrome 15 (1): 93.
Pham, T.K., T.H.T. Nguyen, J.M. Yi, G.S. Kim, H.R. Yun, H.K. Kim, and J.C. Won. 2023. Evogliptin, a DPP-4 inhibitor, prevents diabetic cardiomyopathy by alleviating cardiac lipotoxicity in db/db mice. Experimental and Molecular Medicine 55 (4): 767–778.
Ma, Z.-Y., J. Li, X.-H. Dong, Y.-T. Cui, Y.-F. Cui, T. Ban, and R. Huo. 2023. The role of BRG1 in epigenetic regulation of cardiovascular diseases. European Journal of Pharmacology 957: 176039.
Lin, H., Y. Zhu, C. Zheng, D. Hu, S. Ma, L. Chen, Q. Wang, Z. Chen, J. Xie, Y. Yan, et al. 2021. Antihypertrophic memory after regression of exercise-induced physiological myocardial hypertrophy is mediated by the long noncoding RNA Mhrt779. Circulation 143 (23): 2277–2292.
Funamoto, M., Y. Sunagawa, Y. Katanasaka, K. Shimizu, Y. Miyazaki, N. Sari, S. Shimizu, K. Mori, H. Wada, K. Hasegawa, et al. 2021. Histone acetylation domains are differentially induced during development of heart failure in dahl salt-sensitive rats. International Journal of Molecular Sciences 22 (4): 1771.
Zhang, Y., H. Wang, M. Song, T. Xu, X. Chen, T. Li, and T. Wu. 2020. Brahma-related gene 1 deficiency in endothelial cells ameliorates vascular inflammatory responses in mice. Frontiers in Cell and Developmental Biology 8: 578790.
Yang Yuan, C.W. 2014. Jibin Xu, Jin Tao, Zhiyun Xu, Shengdong Huang: BRG1 overexpression in smooth muscle cells promotes the development of thoracic aortic dissection. BMC Cardiovascular Disorders 14: 144.
Li, Z., X. Zhang, S. Liu, S. Zeng, L. Yu, G. Yang, J. Guo, and Y. Xu. 2018. BRG1 regulates NOX gene transcription in endothelial cells and contributes to cardiac ischemia-reperfusion injury. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1864 (10): 3477–3486.
Chen, B., Q. Zhao, T. Xu, L. Yu, L. Zhuo, Y. Yang, and Y. Xu. 2020. BRG1 activates PR65A transcription to regulate NO bioavailability in vascular endothelial cells. Frontiers in Cell and Developmental Biology 8: 774.
Wang, Y., H. Li, H. Huang, S. Liu, X. Mao, and S. Wang. 2016. Wong Stanley S-c, Xia Z, Irwin Michael G: Cardioprotection from emulsified isoflurane postconditioning is lost in rats with streptozotocin-induced diabetes due to the impairment of Brg1/Nrf2/STAT3 signalling. Clinical Science 130 (10): 801–812.
Tokuyama, T., and S. Yanagi. 2023. Role of Mitochondrial Dynamics in Heart Diseases. Genes 14 (10): 1876.
Mittal, A., R. Garg, A. Bahl, and M. Khullar. 2021. Molecular mechanisms and epigenetic regulation in diabetic cardiomyopathy. Frontiers in Cardiovascular Medicine 8: 725532.
Zhang, X., Y. Lu, J. Wang, and N. He. 2019. Overexpression of Brg1 alleviates high glucose-induced retinal ganglion cell apoptosis though regulating Notch/Hes1 signaling. Biochemical and Biophysical Research Communications 514 (4): 1160–1166.
Liu, M., T. Sun, N. Li, J. Peng, D. Fu, W. Li, L. Li, and W.-Q. Gao. 2019. BRG1 attenuates colonic inflammation and tumorigenesis through autophagy-dependent oxidative stress sequestration. Nature Communications 10 (1): 4614.
Castro, R.O., L. Previato, V. Goitea, A. Felberg, M.F. Guiraldelli, A. Filiberti, and R.J. Pezza. 2017. The chromatin-remodeling subunit Baf200 promotes homology-directed DNA repair and regulates distinct chromatin-remodeling complexes. Journal of Biological Chemistry 292 (20): 8459–8471.
Hopfner, K.-P., and V. Hornung. 2020. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nature Reviews Molecular Cell Biology 21 (9): 501–521.
Sun, Z., and V. Hornung. 2022. cGAS–STING signaling. Current Biology 32 (13): 730–734.
Gamdzyk, M., D.M. Doycheva, C. Araujo, U. Ocak, Y. Luo, J. Tang, and J.H. Zhang. 2020. cGAS/STING pathway activation contributes to delayed neurodegeneration in neonatal hypoxia-ischemia rat model: possible involvement of LINE-1. Molecular Neurobiology 57 (6): 2600–2619.
Luo, W., X. Zou, Y. Wang, Z. Dong, X. Weng, Z. Pei, S. Song, Y. Zhao, Z. Wei, R. Gao, et al. 2023. Critical role of the cGAS-STING pathway in doxorubicin-induced cardiotoxicity. Circulation Research 132 (11): 223–242.
Cheedipudi, S.M., S. Asghar, and A.J. Marian. 2022. Genetic ablation of the DNA damage response pathway attenuates lamin-associated dilated cardiomyopathy in mice. J Am Coll Cardiol Basic Trans Sci 7 (12): 1232–1245.
Hu, S., Y. Gao, R. Gao, Y. Wang, and Y. Qu. 2022. Yang Je, Wei X, Zhang F, Ge J: The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. International Immunopharmacology 107: 108658.
Funding
This work was supported in part by grants from the National Natural Science Foundation of China (No.82104170), the Science and Technology Project of Qingyuan (No.2022KJJH025), the Natural Science Foundation of Guangdong Province (No. 2023A1515010412), the open research funds from Affiliated Qingyuan Hospital, Guangzhou Medical University, Qingyuan People's Hospital (No.202301–208), the Medical Research Fund of Guangdong Province(No.B2023379), the research project of Traditional Chinese Medicine Bureau of Guangdong Province (No.20222299) and the Plan on Enhancing Scientific Research in Guangzhou Medical University (No. GZMU-SH-009/016).
Author information
Authors and Affiliations
Contributions
Participated in research design: XPL, NH, JWZ, ZYC, XML and JXL. Conducted experiments: ZYC, XML, JXL, XY, YLL, XJZ, ZFK and ZZOY. Supervised experiments: XPL, NH and JWZ. Performed data analysis: ZYC, XML, JXL, XY and ZZOY. Contributed new reagents or analytic tools: YLL, XJZ, ZFK and ZZOY. Wrote or contributed to the writing of the manuscript: XPL, NH, JWZ, ZYC, XML and JXL. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethical Standards
The animal procedures were approved by Affiliated Qingyuan Hospital, Guangzhou Medical University (LAEC-2022–015, 19/04/2022) and followed the Guidelines of the Care and Use of Laboratory Animals issued by the Chinese Council on Animal Research.
Conflict of Interest
The authors declare that there are no conflicts of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Lai, X., Li, J. et al. BRG1 Deficiency Promotes Cardiomyocyte Inflammation and Apoptosis by Activating the cGAS-STING Signaling in Diabetic Cardiomyopathy. Inflammation (2024). https://doi.org/10.1007/s10753-024-02058-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10753-024-02058-7