
Abstract
The novel coronavirus SARS-CoV-2, responsible for the COVID-19 outbreak, has become a pandemic threatening millions of lives worldwide. Recently, several vaccine candidates and drugs have shown promising effects in preventing or treating COVID-19, but due to the development of mutant strains through rapid viral evolution, urgent investigations are warranted in order to develop preventive measures and further improve current vaccine candidates. Positive-sense-single-stranded RNA viruses comprise many (re)emerging human pathogens that pose a public health problem. Our innate immune system and, in particular, the interferon response form an important first line of defense against these viruses. Flexibility in the genome aids the virus to develop multiple strategies to evade the innate immune response and efficiently promotes their replication and infective capacity. This review will focus on the innate immune response to SARS-CoV-2 infection and the virus’ evasion of the innate immune system by escaping recognition or inhibiting the production of an antiviral state. Since interferons have been implicated in inflammatory diseases and immunopathology along with their protective role in infection, antagonizing the immune response may have an ambiguous effect on the clinical outcome of the viral disease. This pathology is characterized by intense, rapid stimulation of the innate immune response that triggers activation of the Nod-like receptor family, pyrin-domain-containing 3 (NLRP3) inflammasome pathway, and release of its products including the pro-inflammatory cytokines IL-6, IL-18, and IL-1β. This predictive view may aid in designing an immune intervention or preventive vaccine for COVID-19 in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a member of the genus Betacoronavirus within the family Coronaviridae. It is a single-stranded positive-sense RNA virus. Following the recent outbreak of a novel coronavirus (SARS-CoV-2), COVID-19 has rapidly spread across the globe and has adversely affected the capacity of the global public health community [1, 2]. Among individuals of different age groups, COVID-19 has been reported to cause more severe disease-related symptoms in older age groups and also people with different comorbidities [3], indicating increased disease severity in hosts with weak immune systems. The genome of SARS-CoV-2 (Fig. 1) contains 14 open reading frames (ORFs) which encode 27 different proteins, including the spike (S) protein, envelope (E) protein, membrane (M) glycoproteins, and nucleocapsid (N) protein [4]. Emerging genetic and clinical data of this virus strongly suggests similarities with two previous highly pathogenic human β-coronaviruses, SARS-CoV-1 and MERS-CoV. Approximately, 79% and 50% of sequence identity is seen with SARS-CoV-1 and MERS-CoV, respectively [5]. They also share similar mechanisms for viral entry into the host cells [6] and the propensity to induce hyperinflammation during diseases severity [7]. SARS-CoV-2 infection causes serious respiratory disease similar to SARS-CoV-1, namely, novel coronavirus disease 19 (COVID-19). Common symptoms include fever, cough, shortness of breath, and myalgia or fatigue. Some patients based on ethnicity, age, and genetic makeup display severe symptoms which progress to acute respiratory distress syndrome (ARDS) and multiple organ failure [8]. Despite the identification of this virus, no highly effective antiviral drug has currently been developed as a robust and effective treatment. Fortunately, many vaccine candidates have shown promising results, including prevention of developing severe symptoms and substantially reducing hospitalizations of infected people. However, the long-term efficacy of vaccines and mechanisms exacerbating the disease still largely remain undetermined. The innate immune system relies mainly on different pattern recognition receptors (PRRs) to detect pathogen-associated molecular patterns [8]. Recognition of viral genome during infection plays an important role in limiting virus replication at the early stages of infection. During the process, the Nod-like receptor family, pyrin domain-containing 3 (NLRP3), is activated [9]. Currently, there is a gap in knowledge regarding the host innate immune response to SARS-CoV-2. However, based on the accumulated clinical and experimental data on these previous viruses, predictions can be made on how the host immune system may deal with this virus and how the virus may evade such host responses [10]. The first line of defense against viral infection comprises a set of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), that recognize the RNA viral genome and its replication intermediates. Evidence suggests that upon entry into the alveolar epithelium, the virus is sensed by the endosomal single-stranded (ss) RNA sensor, TLR7/8, and the cytosolic double-stranded (ds) RNA sensor, RIG-I/MDA-5. Upon recognition, these sensors recruit the adaptor proteins, MyD88 and MAVS, respectively, and induce downstream signaling. Ultimately, this leads to the activation of the transcription factors, IRF3/7 and NF-κB, and the subsequent production of type I interferons (IFN-α and IFN-β) and pro-inflammatory cytokines (e.g., IL-6 and TNF-α), respectively [11].

Pictures depicting the structure organization and ORFs of SARS-CoV2. A Structure of SARS-CoV2: it is an enveloped virus, nonsegmented with positive‐sense single‐stranded RNA genome. Various components of the virion are shown as follows: spike protein (S), the envelope (E) protein together with the membrane (M) protein, and the nucleocapsid (N) protein bound to the RNA genome forming nucleocapsid. B Graph representing genomic organization of SARS-CoV2: genome of the virus encodes two large open reading frames (ORF1a and ORF1b) as shown in 5′ to 3′ orientation. About two-thirds (67%) of the complete virus genome consist of two ORFs: ORF1 a and b. Both these ORFs encode for 16 nonstructural proteins. The remaining ORFs occupy the remaining one-third of the genome encoding the four structural proteins (S, spike; E, envelope; M, membrane; N, nucleocapsid) and other accessory proteins of the virus.
NLRP3 inflammasome is an oligomeric complex comprised of the NOD-like receptor NLRP3, the adaptor ASC, and caspase-1. This complex is crucial for generating optimal antimicrobial response in host body accompanied with IL-1β and IL-18 secretion and pyroptosis. NLRP3 recognizes different varieties of PAMPs and danger-associated molecular patterns (DAMPs) during viral infection, triggering NLRP3 inflammasome-dependent antiviral immune response to remove virus. However, viruses have evolved effective ways in evading the immune system, such as through modulating the NLRP3 inflammasome. Indeed, SARS-CoV-1 has previously been shown to induce the formation of the NLRP3 inflammasome through the action of viral proteins such as the E and 3a proteins [12]. However, our understanding of the exact innate immune viral recognition mechanisms requires a further understanding. Subsequently, our paper aims to provide a conceptual knowledge of host–pathogen interactions relevant to the innate immune system. It also suggests future avenues pertaining to the treatment of COVID-19.
INNATE IMMUNE SENSING OF SARS-COV-2
Innate immune system in animals consists of conserved cellular and molecular defense strategies involved in recognition and removal of pathogens and also sending signals for activation of adaptive immune response. Against intracellular pathogens, innate immunity acts as a first line of defense. Innate immune response generation involves recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognizing receptors expressed by host cells (PRR) [13]. Interaction of PAMP with PRR triggers a signaling response, which leads to the synthesis and secretion of battery soluble cytokines. Understanding of the specific innate immune response against SARS-CoV-2 is currently limited. However, based on our existing knowledge of other CoVs, we can recapitulate common mechanisms during SARS-CoV-2 interactions with host cells.
During SARS-CoV-2 infection, viral spike protein interacts with ACE2 receptor on the surface of immune cells [14], and the viral RNAs inside the infected cell detect intracellular PRR, resulting in immune system activation (Fig. 2). RNAs of viruses including SARS-CoV, SARS-CoV-2, and MERS-CoV are detected by endosomal RNA detecting PRRs, including Toll-like receptors (TLR-3 and 7) and/or cytoplasmic RNA sensors, namely, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) [15] (Fig. 2). During the process of viral RNA recognition inside immune cells, the downstream signaling complex is activated, ultimately leading to activation of crucial factors involved in generating antiviral signaling pathways. Key factors involved in the process are IRF3 (IFN regulatory factor-3), nuclear factor κB (NF-κB), and JAK (Janus kinase)/STAT (signal transducer and activator of transcription) [16]. These cascades during infection are important for diseases progression, and therefore, identification of complete signaling molecules in the pathway may serve as potential therapeutic targets. In line with this, pathways activated by TLRs have been found to differentially control other different signaling pathways in human CD14 + monocytes. Secretion of specific cytokines and type I IFN takes place by coordinated activity of signaling pathways [15].

Schematic representation of elements involved in generating innate immune response against SARS-CoV-2. SARS‐CoV2 infects permissible cells via angiotensin‐converting enzyme 2 (ACE2). After entering the alveolar epithelium, the virus is recognized by important innate immune sensors including endosomal RNA sensors—Toll-like receptor 3 (TLR3), TLR7/8 and cytoplasmic retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated protein 5 (MDA5). TLRs are known to further activate TIR-domain-containing adapter-inducing IFNβ (TRIF) and myeloid differentiation primary response gene 88 (MyD88) signaling pathways. MyD88-dependent pathway proceeds via formation of “Mydossome” with TRAF6 activating TAK1 kinase via polyubiquitination. The activated TAK1 activates IKK kinase complex by phosphorylation. The activation of IKK complex leads to the activation of induced nuclear translocation of NF-κB. The TRIF-dependent pathway, on the other hand, recruits TRAF3 and TRAF6. TRAF3 activates IKK complex by polyubiquitination which in turn activates IKKε/TBK1 by phosphorylation and causes activation and nuclear translocation of IRF3 and IRF7. In addition, virus-induced mitochondria damage activates cyclic GMP-AMP synthase (cGAS) and stimulator of interferon gene (STING) pathway to induce synthesis of antiviral IFN-α/β production via IRF3 and IRF7. Transcription factor NF-κB initiates production of pro-inflammatory cytokines (TNF-α and IL-6), and the transcription factor IRF3/7 initiates production of type I interferon (IFN-α/β). Interferons are secreted and bind to the type I interferon receptor (IFNAR) in an autocrine loop to activate JAK-STAT signal transduction pathway, where STAT1-STAT2 is phosphorylated and forms heterodimer that joins IRF9 to form ISGF-3. ISGF-3 complex then binds to ISREs on the regulatory region on target genes to induce expression of interferon-stimulated genes (ISGs). ISG genes expression establishes an antiviral state in the cells.
Usually, TLRs upon recognition trigger two independent but related pathways: the first one depends upon the recruitment of the adapter protein myeloid differentiation primary response gene 88 (MyD88), and the second one proceeds via the adapter TIR-domain-containing adapter-inducing IFN-β (TRIF) [17]. TLR3 signaling is accompanied with the recruitment of TRIF, which further interacts with tumor necrosis factor receptor-associated factors 3 and 6 (TRAF3 and TRAF6). This signaling complex further activates a serine/threonine-protein kinase (TBK1) and inhibitor of κB kinase (IKK) and stimulating nuclear factor κ enhancer light chains of activated B cells (NF-κB), IFN regulatory factor (IRF) 3, and cytokine secretion. Additionally, TRAF6 also interacts with RIP, leading to stimulation of transforming growth factor beta-activated kinase 1 (TAK-1) complex and activation of NF-κB and mitogen-activated protein kinase (MAPK) [18]. During TLR7 engagement, MyD88 is involved and forms a scaffold complex with IL-1 receptor-associated kinases 1 and 4 (IRAK1 and IRAK4), TRAF3, and TRAF6. These multiprotein complexes stimulate MAPK, IKK, and TBK, which downstream activates and promotes nuclear translocation of activator protein 1 (AP-1), NF-κB, IRF3, and IRF7. The coordinated activity of all these transcription factors results in induction of IFN-I, certain pro-inflammatory cytokine, and chemokine (Fig. 2) [18, 19].
Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are additional RNA sensor molecules located in the cytoplasm of various cells, including myeloid, epithelial, and central nervous system. These sensor molecules are involved in sensing phosphate-containing RNA and long dsRNA to provide antiviral immunity [20]. Following virus genome recognition, the RLRs induce a downstream signaling response and interact with mitochondrial membrane and activate mitochondrial antiviral signaling (MAVS) and then activate transcription factors such as interferon regulatory factor 1 (IRF1), IRF3, and NF-κB to activate the synthesis of types I and III interferons and inflammatory cytokines [21]. RLRs induced signaling pathway has been reported to result in an aberrant cytokine storm condition that leads to debilitating conditions in COVID-19 infected patients [22].
Type I IFNs (IFN-α/β) bind to their specific IFN-α/β receptors (IFNARs) on the surface of nearby or viral infected cells, activating the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway to synthesize ISGs for promoting antiviral activity [23]. Detailed signaling process for type I IFNs signaling involves activation of JAK1 and tyrosine kinase 2, causing phosphorylation and dimerization of STAT1 and STAT2 [24], which later forms a complex with IRF9. STAT-IRF-9 complex translocates to the nucleus to induce transcription of ISGs [25]. ISGs are known to influence several key cellular processes, including but not limited to RNA processing, protein stability, and cell viability, which directly participates in the viral life cycle. ISGs are also involved in activating T and B cell [26], thereby affecting the magnitude and resolution of adaptive immune response for virus clearance.
Other than IFNs, one notable activator of JAK/STAT signaling pathway is IL-6, which induces biological effects during infection. IL-6 has been reported to increase exponentially COVID-19 patients [27,28,29] with a strong implication in acute inflammation and cytokine storm. The main targets of IL-6 are T cells, B cells, and many granulocytes. It is known to control activation, differentiation, and survival of B and T cells. During SARS-CoV-2 infection, large amounts of IL-6 are produced and released by fibroblast, mesenchymal, endothelial, and other cells. This leads to the hyperactivation of T cells causing fatal immune reactions in patients. This pathological release of IL-6 results in severe symptoms in infected individuals.
NLRs (NOD-like receptor) are another large family of cytosolic proteins which functions as PRR for generating response against viruses. This class of receptors is organized into three main domains: a CARD domain, pyrin domain (PYD), or baculovirus inhibitor repeat domain at the N terminal, a conserved NOD motif at the intermediate region and LRR motifs at the C terminal. The LRR motifs of the receptor detect viral PAMPs and subsequently activate MAPK and NF-κB signaling pathways [30]. Furthermore, some members of NLRs are known to generate multimeric protein complexes known as inflammasomes. These inflammasomes are mainly responsible for preventing survival of pathogens inside the infected cells and also induce the cleavage of pro-IL-1β and pro-IL-18 into their active forms [31].
The transcription factor NF-κB is a major regulator for both innate and adaptive immunity [32]. It is essential in initiation and propagation of optimal immune responses and, therefore, participates in decreasing inflammation [33]. However, overstimulation of the NF-κB signaling pathway often results in the development of inflammatory diseases (Hayden et al. 2006) [33]. Notably, the exacerbation of NF-κB activation is also reported to have implications in lung inflammatory immunopathology induced by respiratory viruses like SARS-CoV [34]. In line with this, [35] demonstrated that stimulation of murine macrophages cell line (RAW264.7) with recombinant SARS-CoV-2 spike protein resulted in excessive secretion of IL-6 and TNF-α in a dose-dependent manner. Synthesis of both cytokines relies on the activation of NF-κB signaling pathway [36]. Accordingly, transfection of RAW264.7 cells with dominant negative NIK which inhibits NF-κB activation was seen to produce a blunted secretion of IL-6 and TNF-α upon spike protein stimulation [36].
The cyclic GMP-AMP synthase (cGAS) and stimulator of IFN gene (STING) pathway are also activated during viral infection [37]. This pathway involves recognition of pathogen DNA and also self-DNA. In the process, the cGAS nucleotidyl transferase produces second-messenger cyclic GMP-AMP (cGAMP) that binds to STING, leading to activation of TBK1, phosphorylation of IRF3, and production of type I IFNs [38]. Overactivation of STING has been shown to induce progressive activation of CD4 + and CD8 + T lymphocytes as observed in severe COVID-19 patients. Targeting STING pathway with specific inhibitors could be regarded as a therapeutic strategy for treating severe COVID-19 patients.
MODULATION OF HOST INNATE IMMUNITY BY SARS-COV-2
Based on recent literature, coronaviruses have particularly been shown to adapt to evade immune detection whilst also compromising host immune responses. This partially explains the longer incubation period of such viruses, 2–11 days on average compared to other common flu virus like influenza, which displays an incubation period of 1–4 days [39]. The length of incubation period is determined by several factors such as the number of viral particles infecting the host and how efficiently they are cleared by host immunity and viral evasion strategies. The longer incubation period for CoVs is, therefore, due to more efficient immune evasion strategies at early stages of infection.
Based on findings from HCoVs, most likely PRRs involved in SARS-CoV-2 sensing are TLR3, TLR7, RIG1, and MDA5 as discussed previously [40]. And also, all these PRRs induce signaling cascades for inducing very strong IFN response. Previous reports have shown that infection caused by HCoVs, especially the highly pathogenic SARS-CoV-1, suppresses IFN release in vitro and in vivo [41, 42]. In line with this, SARS-CoV-2 also displayed weak IFN I/III signatures from infected cell lines, primary bronchial cells, and a ferret model [43]. In fact, severe COVID-19 patients show remarkably impaired IFN I signature when compared with mild or moderate cases [44, 45]. The capability of the virus to control I IFN signaling is an important feature of its virulence properties [46]. The complexities are associated with IFN synthesis and secretion; it is utmost essential to understand the complete dynamics of type I IFN response during SARS-CoV-2 infection. Different cell lines model demonstrates SARS-CoV-2-induced low level of type I and II IFNs response, ultimately inducing moderate amount of ISGs and key pro-inflammatory signature molecules including ILIB, IL6, TNF, and different chemokines [43]. An early clinical study reported that type I IFNs were either not detected or found in very small amount irrespective of disease severity in the plasma of patients [44]. These findings suggest that early burst of type I IFN (IFN-α) is more likely of lung origin rather than derived from blood-derived immune cells [47]. The dynamics of type I IFNs in mouse model suggest that they play a significant role as a driver of pathologic response [48].
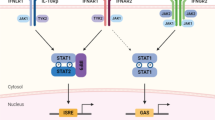
SARS-CoV-2 uses a multipronged approach to evade type I innate immune response which mainly relies on the viral components and their interactors in host cells in early COVID-19 studies [40, 43, 47]. At least ten SARS-CoV-2 proteins have been identified that counteract the antiviral action of IFN (Fig. 3).

Immune evasion strategies exploited by SARS-CoV2 during infection. After viral genome entry into the host cells during infection, viral genome ssRNA as well as dsRNA intermediate found in virus life cycle is sensed by innate immune sensors, RIG-I/MDA5 in cytoplasm or Toll-like receptors TLR3/7/8 in endosome. Response generated from these sensors initiates a downstream signaling cascade leading to IFN-β gene expression. RIG-I/MDA5-dependent signaling involves a mitochondrial adaptor MAVS, whereas TLR signals through TRIF/MyD88. Both pathways involve common TRAF adaptor to activate transcription factors. The SARS-CoV-2 encoded proteins shown in yellow box are known to intervene the host innate immune signaling at various action points as evasion mechanisms to sustain viral replication and propagation. One key strategy is to effectively suppress the activation of TNF receptor-associated factors (TRAF) 3 and 6, thereby limiting activation of the transcription factors NFκB and IRF3 and 7. This leads to severely dampened early pro-inflammatory response mediated by type I interferons (IFN) and pro-inflammatory effector cytokines IL-1, IL-6, and TNF-α. Furthermore, novel SARS-CoV-2 inhibits activation of STAT transcription factors (ISRE) in response to type I IFN receptor activation, which further limits antiviral response mechanisms. Altogether, this prohibits virus containment through activation of antiviral programs and the recruitment of immune cells.
A nonstructural protein 16 (NSP16) of SARS-CoV-2 suppresses global mRNA splicing and viral mRNA sensing by host helicase receptors. In addition to this, NSP1 causes global inhibition of mRNA translation, and NSP8 and NSP9 interfere with protein movement to the cell membrane. All these processes ultimately lead to reduced type I IFN production by infected cell [49, 50]. Biophysical analysis further suggested that NSP13 interacts with TBK1 and NSP15 with NRDP1 (E3 ubiquitin ligase for maintaining JAK-STAT receptor degradation). Other viral proteins including ORF9b and ORF6 were seen to interact with TOMM70 (outer mitochondrial membrane protein) and KPNA2 or IFN-induced NUP98-RAE nuclear export complex, which are both involved in nuclear targeting of proinflammatory factors such as IRF3, IRF7, and STAT1 [50, 51].
Functional testing demonstrated further support for the counteracting activity of ORF3b, ORF6, NSP1, or NSP13 on type I IFN activation, of which particularly ORF6 suppressed STAT1 and STAT2 phosphorylation and STAT1 nuclear translocation [51, 52]. Evidence suggests that SARS-CoV-2 is targeting the type I IFN system at various steps, subsequently strongly interfering with a well-orchestrated interplay between antiviral and pro-inflammatory innate and adaptive defense mechanisms within the immune system. Early studies of COVID-19 infection, especially associated with severe disease, suggested infection-induced immune response results to cytokine storm or cytokine release syndrome (CRS) [53, 54]. In severe or critical patients, NF-kB pathway was seen to be mainly involved for upregulated expression of several pro-inflammatory genes [44]. This response later leads to accumulation of pathogenic inflammatory neutrophils and macrophages in the lung, further causing higher load of pro-inflammatory cytokines and chemokines in BALF and blood [1, 35, 43, 44, 55,56,57]. Among cytokines, IL-6, TNF, and IFNγ have shown elevated responses in mild to severe patients [44, 58]. Among these, longitudinal profiling revealed dynamic changes in the concentration of IL-6 correlates with the pathology of COVID-19. Collectively, these findings further support more complex viral-host interactions with upregulation of pro-inflammatory signaling as a crucial part of derailed innate immune response.
ALTERATIONS IN INNATE IMMUNE CELLS DURING SARS-COV-2 INFECTION
Series of cellular and transcriptional changes take place in the respiratory tract to generate mucosal immune response against SARS-CoV-2 infection [59]. As expected, very large influx of innate immune cells take place in the lungs of COVID-19 infected patients. Most notable cells involve neutrophils and macrophages attracted toward lungs epithelium in response to several chemokines secreted by infected lung epithelial cells. Due to the hyperactivity of pro-inflammatory macrophages, further recruitment of more granulocytes and monocytes takes place in patients showing severe symptoms [59, 60]. It is still not clear whether changes seen in the immune cells in circulation reflect individual-patient changes in lungs with known viral load and infection. One of the key hallmarks of severe COVID-19 involves an increase in circulating neutrophils along with decreased proportions of lymphocytes, mainly clonally expanded CD8 + T cells [1, 44, 61, 62]. Furthermore, severe patients show dysfunctional S100hi classical monocytes with downregulated MHC-II expression, ultimately promoting sepsis [47, 63,64,65]. In addition to this, IFN-α secreting plasmacytoid DCs (pDCs) are both reduced and functionally impaired in the circulation of infected patients, meaning infected lungs act as an important source for elevated levels of type I IFNs during early stages of infection [47, 66]. As discussed early, lymphopenia is identified as an important hallmark of severity of disease which not only includes reduction in B and T cells but also in NK cells [44, 67] Patients with faster recovery are accompanied with decreased inhibition of NF-κB signaling in NK cells along with increased levels of cytotoxicity to clear viral infected cells more efficiently [63]. All in all, immunosuppressive phenotypes in innate immune cells are directly linked to deviation in the adaptive immune systems observed during SARS-CoV-2 infection, which might be leading to lymphopenia [62]. Overall, we postulate that very much different molecular determinants might be involved in differently programming innate immune cells in mild versus severe disease courses, accompanied with differences in adaptive immunity against SARS-CoV-2. This is sufficient to further understand inflammasone signaling pathway during viral infection which might be a converging point toward a detrimental clinical path.
NLRP3 INFLAMMASOMES ACTIVATION IN SARS-COV-2 PATHOGENESIS
The NLRP3 is a vital sensor protein in cytosol involved in producing antimicrobial response. It contains an N-terminal pyrin domain (PYD), a central NACHT domain, and C-terminal leucine-rich domain (LRR) [68]. During response, NLRP3 oligomerizes and causes cleavage-dependent activation of caspase 1, which ultimately leads to inflammasome activation [69]. Upon activation, inflammation is enhanced by increased production of molecules including IL‐1β, IL‐18, and gasdermin D which plays a key role in regulating pathogen-induced inflammation. Numerous studies have shown excessive and aberrant inflammasome activity during viral infection, ultimately causing systemic inflammation-induced tissue destruction. In addition to this, NLRP3 inflammasome and IL-1β are known to mediate inflammation during lung injury and ARDS [70, 71]. Recent studies on SARS‐CoV‐2 using wide range of cells indicate activation of the inflammasome pathway [72]. It is more likely that NLRP3-mediated inflammasome is regarded as a major cause of formation of the severe inflammatory cytokine storm, resulting in clinical and pathological manifestations of individuals infected with COVID-19 [73]. In other coronavirus infections including MERS-CoV and SARS-CoV-1, patients with ARDS show high levels of pro-inflammatory markers such as IL-1β, IL-6, and IL-8 [73, 74]. In line with this, influenza infection is known to demonstrate a high load of IL-1β in the bronchoalveolar fluid and plasma from patients with lung injury [75,76,77,78,79]. Based on these reports, IL-1β appears to play a key role in acute lung injury, so subsequently, pharmacological targeting of this pathway represents an important area of intervention. The acute immune response generated during SARS-CoV-2 infection is largely driven by inflammatory alveolar and monocyte-derived macrophages in response to PAMPs and DAMPs sensing in the infected cells [80,81,82]. As an initial response to infection, TNF-α and IL-1β secreted by alveolar macrophages generate an acute pro-inflammatory environment. The secretion of these cytokines induces cell death and damage, PAMP/DAMP production, immune cell recruitment, and widespread NLRP3 activation, establishing a pro-inflammatory positive feedback cascade [82,83,84]. Through the aid of ZBP1, NLRP3 recognizes viral proteins and promotes inflammasome assembly [85]. ZBP1 interacts with the IAV nucleoprotein (NP) and polymerase subunit PB1 after infection. ZBP1 goes on to interact with RIP3 through their shared domain homotypic interaction motif (RHIM), to stimulate activation of the NLRP3 inflammasome via the RIP1-RIP3-caspase-8 pathway [85,86,87].
During the course of infection, viruses stimulate various changes in cellular status of their host cells, including lysosomal maturation, aberrant ion concentrations, mitochondria damage, and the accumulation of misfolded protein aggregates, all of which are recognized as danger signals by the host and stimulate the activation of the NLRP3 inflammasome. The maturation and acidification of lysosomes cause the leakage of catalytically active cathepsin B, and the resulting generation of reactive oxygen species (ROS), which activates the NLRP3 inflammasome [88, 89]. ROS are also required for NLRP3 inflammasome activation. Potassium efflux is a well-known activator of the NLRP3 inflammasome [90, 91]. N protein from SARS-CoV causes the flux of calcium from intracellular storages to the cytosol, which is vital for NLRP3 activation [92]. SARS-CoV viroporin 3a changes membrane permeability through formation of a cation-selective ion channel. Subsequently, the ion channel permits the release of Na + /K + , rather than Ca2 + , to induce the NLRP3 inflammasome activation [12]. Mitochondria damage is also a vital activator of the NLRP3 inflammasome. Similar to lysosomal or endosomal maturation, mitochondria damage also leads to the production of ROS to activate the NLRP3 inflammasome [93].
The SARS-CoV-1 genome encodes 3 ion channel proteins: E, open reading frame 3a (ORF3a), and ORF8a in which E and ORF3a are required for both replication and virulence [83, 94, 95]. In addition to the canonical NLRP3 activation pathway by PAMPs and DAMPs, the E, 3a, and 8b proteins of SARS-CoV-1 function as NLRP3 agonists [81, 83, 96]; many of these sequences are conserved in SARS-CoV-2 and likely play a role in inflammatory pathogenesis [81, 97].
Defining the inflammatory activities of SARS-CoV-2 ORFs 3a and 8 is therefore critical to predictive monitoring and modeling of novel SARS-CoV-2 strain emergence.
The NLRP3 inflammasome has demonstrated to play an instrumental role in the pathogenesis of viral diseases [98, 99]. The proliferation of SARS-CoV-2 in various cells can be combined with numerous observations of direct and indirect activation of inflammasome by other coronaviruses. Activation of the inflammasome is likely to be involved in the formation of severe cytokine storm, which subsequently causes ARDS and MODS and ultimately leads to death. Recent studies emphasize on the key role of NLRP3 inflammasome in immunopathogenesis of severe COVID-19 especially in patients with increased risk (such as diabetes and obesity) [100,101,102].
For SARS-CoV-2-induced NLRP3 inflammasome activation, the potential mechanisms are based mainly on canonical activation pathways. SARS-CoV-2 can activate the NLRP3 inflammasome either directly or through diverse cellular/molecular signaling events. Incubation of viable SARS-CoV-2 viral particle with monocytes causes the activation of NLRP3 inflammasome as demonstrated by puncta formation, a marker for active inflammasome formation. Activation of NLRP3 inflammasome is associated with disease severity in COVID-19 patients [103].
A number of hypotheses suggest that several proteins encoded by coronavirus, called viroporins, are responsible for ion channel proteins (IC) assemblage and alteration of cell membrane permeability, facilitating virus spreading and virulence [104, 105]. The accessory protein 3a of SARS-CoV-2 contains several functional domains that have been suggested to be involved in the virus’ virulence and pathogenesis by the activation NLRP3 inflammasome [106]. Given its capacity to interact directly with both transcription factor TNF receptor 3 (TRAF3) (through domain II) and ASC NLRP3 protein (through domain III) and enhance IC assemblage (through domain III), 3a acts as an important factor in the activation signaling pathways of NFk-B and pro-cytokine maturation to activate IL-1β and IL-18 [106, 107].
The N proteins of SARS-CoV-2 can activate the complement cascade in a mannan-binding lectin-dependent manner. Several studies have indicated that the initiation of the complement cascade can lead to the activation of the NLRP3; the complement system contributes to COVID-19 pathology and is closely related to the activation of the NLRP3 inflammasome. In this context, it can be inferred that SARS-CoV-2 infection activates the NLRP3 inflammasome through the complement cascade pathway [108, 109].
Infection with SARS-CoV-2 induces cell death, characterized by loss of integrity of the plasma membrane, characteristic of pyroptosis. Evaluation of COVID-19 patients and postmortem samples showed that SARS-CoV-2 induces inflammasome activation in primary human monocytic cells and mimics the release of lactate dehydrogenase, a marker of cell injury, from infected monocytes. According to recent reports, SARS-CoV-2 directly infects human monocytic cells and promotes activation of NLRP3 and lytic cell death [22, 110]. Although further evidence is needed to verify the role of both structural and nonstructural SARS-CoV-2 proteins, understanding the underlying molecular mechanisms could pave the way for a therapeutic target to reduce disease severity (Fig. 4).

NLRP3 activation by SARS-CoV-2. Activation of the NLRP3 inflammasome requires priming signal and activation signal. During initial priming signal, activated PRRs induce IRF3/7 and NF-kB activation, triggering the transcription of NLRP3, pro caspase-1, pro-IL-1b, and pro-IL-18. Later, activation signal involves multiple DAMPs, and PAMPs induced NLRP3 inflammasome assembly and activation. DAMPs include lysosomal or endosomal injury, aberrant ionic fluxes, mitochondrial injury, and protein aggregates. With the help of ZBP1, NLRP3 is activated by sensing viral proteins and RNA and promotes inflammasome assembly. Lysosomal maturation and mitochondria damage induce the production of ROS to activate the NLRP3 inflammasome. NLRP3 recruits the adapter protein, ASC and the protease caspase-1. Association with these factors makes NLRP3 fully matured, and NLRP3 inflammasome further drives maturation of pro-IL-1β and pro-IL-18 into their respective active forms. Activation of caspase-1 involves the auto-cleavage of pro-caspase-1 and matured caspase-1 and then mediates proteolytic cleavage-based activation and secretion of pro-IL-1b and pro-IL-18 into IL-1β and IL-18 which results in pyroptosis (programmed cell death).
EMERGENT TREATMENTS AGAINST COVID-19
Currently, the fundamental tools being used to respond against the global threat posed by COVID-19 are various vaccines. Through a collaboration of governments, pharmaceutical organizations, and academics across the world, several decades of progress on new vaccine platforms, viral immunology, structural biology, protein engineering research, and clinical trial operations expertise allowed rapid development, evaluation, manufacturing, and deployment of successful vaccines [111].
Modern vaccinology, as seen in the vaccines produced by companies such as Pfizer and Moderna, utilizes advances in viral immunology, structural biology, and novel vaccine platforms to safely elicit robust immunity [111]. Recognizing the importance of stabilizing viral surface proteins as immunogens enabled investigators to build their understanding of the atomic-level structure of the HKU1 Betacoronavirus. This allowed investigators to subsequently assess the impact of introducing stabilizing mutations into the spike protein of HKU1, SARS, and Middle East Respiratory Syndrome (MERS) [112, 113]. Both of these studies [112, 113] demonstrated how the alteration of two sequential amino acids to prolines in the central helix of the transmembrane portion of the protein could aid in the stabilization of the spike protein in its native conformation, enhancing protein expression and thus improving immunogenicity [114]. The sequence alignment allowed the two proline mutations to be introduced into the SARS-COV-2 spike protein to facilitate the successful design and production of some of the vaccines which are successfully combating COVID-19 today [111].
However, further research must be conducted upon potential drugs which can combat COVID-19; as to date, dexamethasone is the only drug that has shown to reduce COVID-19 mortality in those requiring respiratory support [115].
CONCLUSION
The COVID-19 pandemic, caused by novel SARS-CoV-2, is one of the greatest global public health emergencies since the Spanish influenza outbreak of 1918. This has left us with an unprecedented challenge for the identification of both preventive and therapeutic drugs. Knowledge of virus interactions with host cells has gained importance for designing effective therapeutics for the treatment. These insights are explored for urgently designing several vaccines and drugs in countering the pandemic. Currently, understanding of the mechanistic detail of host innate immune response remains limited, and further research is required to enhance this understanding. It is highly crucial to understand the host innate immune responses during infection and how pathogens exploit them for successful evasion. One classical and key innate immune response involves antiviral type I IFNs, but several ssRNA viruses show different mechanisms to antagonize IFN response, thereby protecting themselves from host immune regulators. Additionally, pathology is caused through viral evasion associated with a delayed aberrant IFNs induced inflammatory response.
SARS-CoV-2 enters the host cells through binding to cellular receptors, ACE2. After entry into the host cell, the innate immune sensors, TLR and RLR, are activated. Response generated by these regulators further activates IRF3, IRF7, and NF-κB, leading to generation of an antiviral response and the production of inflammatory cytokines. Also, COVID-19 causes an array of disease manifestations, the most severe of which is mediated by a massive inflammatory response which may involve stimulation of the NLRP3 inflammasome. Direct scientific data linking the NLRP3 inflammasome and SARS-CoV-2 infection is limited due to poor understanding of this new pathogen.
However, recent studies directly support the role of IL-1β and NLRP3-dependent inflammasome activation in the pathogenesis of acute lung injury. The potential involvement of NLRP3 in severe cases of COVID-19 warrants further research into the therapeutic targeting of the NLRP3 inflammasome. Overall, more understanding and investigations of the host innate immune response and pathogenesis of SARS-CoV-2 would facilitate more insights for the development of antiviral therapeutics and vaccines.
Availability of Data and Materials
This article has no additional data.
References
Huang, C., et al. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395: 497–506.
Nagaraja, S., et al. 2022. Inflammasome regulation in driving COVID-19 severity in humans and immune tolerance in bats. Journal of Leukocyte Biology 111: 497–508.
Chen, N., et al. 2020. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 395: 507–513.
Wu, A., et al. 2020. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host & Microbe 27: 325–328.
Lu, R., et al. 2020. Genomic characterization and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395: 565–574.
Hoffmann, et al. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 18: 271–280.
Xu, X.W., et al. 2020. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARSCov-2) outside of Wuhan, China: retrospective case series. The BMJ 368: m606.
Medzhitov, et al. 2001. Toll-like receptors and innate immunity. Nature Reviews Immunology 1: 135–145.
Bauernfeind, et al. 2011. Inflammasomes: Current understanding and open questions. Cellular and Molecular Life Sciences 68: 765–783.
Prompetchara, et al. 2020. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific Journal of Allergy and Immunology 38: 1–9.
Lim, et al. 2016. Human coronaviruses: a review of virus-host interactions. Diseases 4.
Chen, I.Y., et al. 2019. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Frontiers in Microbiology 10: 50.
Chen, N., et al. 2017. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 69: 297–304.
Ashraf, U.M., et al. 2021. SARS-CoV-2, ACE2 expression, and systemic organ invasion. Physiological Genomics 53: 51–60.
De Marcken, M., et al. 2019. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Science Signaling 12: eaaw1347.
Olejnik, J., et al. 2018. Toll-like receptor 4 in acute viral infection: too much of a good thing. PLoS Pathog 14: e1007390.
Zhu, J., et al. 2020. Infectious bronchitis virus inhibits activation of the TLR7 pathway, but not the TLR3 pathway. Archives of Virology 165: 2037–2043.
Kawasaki, T., and T. Kawai. 2014. Toll-like Kawai receptor signaling pathways. Front Immunol 5:461 Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M Jr, 2006 Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. Journal of Virology 80: 9424–9434.
Zhou, H., et al. 2013. IRAK-M mediates Toll-like receptor/IL-1R-induced NFkappaB activation and cytokine production. EMBO Journal 32: 583–596.
Loo, Y.-M., and M. Gale. 2011. Immune signaling by RIG-I-like receptors. Immunity 34: 680–692.
Seth, R.B., et al. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122: 669–682.
Li, H., Y. Wang, M. Ji, F. Pei, Q. Zhao, Y. Zhou, Y. Hong, S. Han, J. Wang, Q. Wang, Q. Li, and Y. Wang. 2020. Transmission routes analysis of SARS-CoV-2: A systematic review and case report. Front Cell Dev Biol. 10 (8): 618.
Morrison. J., and A. García-Sastre. 2014. STAT2 signaling and dengue virus infection. JAKSTAT 3: e27715.
Qin, S., et al. 2021. Analyzing master regulators and scRNA-seq of COVID-19 patients reveals an underlying anti-SARS-CoV-2 mechanism of ZNF proteins. Brief Bioinformation 27: bbab118.
Mu, J., et al. 2020. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov 6: 65.
Wani, S.A., et al. 2019. Contrasting gene expression profiles of monocytes and lymphocytes from peste-des-petits-ruminants virus infected goats. Frontiers in Immunology 10: 1463.
Mehta, P., et al. 2020. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033–1034.
Zhang, C., et al. 2020. The cytokine release syndrome (CRS) of severe COVID-19 and interleukin-6 receptor (IL-6R) antagonist tocilizumab may be the key to reduce the mortality. International Journal of Antimicrobial Agents 55: 105954.
Carvalho, T. 2021. The first 12 months of COVID-19: A timeline of immunological insights. Nature Reviews Immunology 21: 245–256.
Kanneganti, T.-D. 2010. Central roles of NLRs and inflammasomes in viral infection. Nature Reviews Immunology 10: 688–698.
Guo, H., et al. 2015. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nature Medicine 21: 677–687.
Li, Q., and I.M. Verma. 2002. NF-kappaB regulation in the immune system. Nature Reviews Immunology 2: 725–734.
Hayden, M.S., et al. 2006. NF-κB and the immune response. Oncogene 25: 6758–6780.
De Diego, M.L., et al. 2014. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. Journal of Virology 88: 913.
Wang, X., et al. 2020. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion.” Cellular & molecular immunology 7: 1–3. https://doi.org/10.1038/s41423-020-0424-9.
Wang, W., et al. 2007. Up-regulation of IL-6 and TNF-alpha induced by SARScoronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Research 128: 1–8.
Sun, B., et al. 2017. Dengue virus activates cGAS through the release of mitochondrial DNA. Science and Reports 7: 3594.
Uno, N., and T.M. Ross. 2018. Dengue virus and the host innate immune response. Emerg Microbes Infect 7: 167.
Lessler, J., et al. 2009. Incubation periods of acute respiratory viral infections: A systematic review. The Lancet Infectious Diseases 9: 291–300.
Sa Ribero, M., et al. 2020. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathogens 16: e1008737.
Cameron, M.J., et al. 2012. Lack of innate interferon responses during SARS coronavirus infection in a vaccination and reinfection ferret model. PLoS One 7: e45842.
Minakshi, R., et al. 2009. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 4: e8342.
Blanco-Melo, D., et al. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181: 1036–1045.
Hadjadj, J., et al. 2020. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369: 718–724.
Lowery, S.A. 2021. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host & Microbe 29: 1052–1062.
Randall, R.E., and S. Goodbourn. 2008. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. Journal of General Virology 89: 1–47.
Arunachalam, P.S., et al. 2020. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369: 1210–1220.
Israelow, B., et al. 2020. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. Journal of Experimental Medicine 217: e20201241.
Banerjee, A.K., et al. 2020. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183: 1325-1339.e21.
Gordon, D.E., et al. 2020. A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. Preprint. bioRxiv 03.22.002386.
Xia, H. et al., 2020. Evasion of type I interferon by SARS-CoV-2. Cell Reports 33: 108234.
Lei, X., et al. 2020. Activation and evasion of type I interferon responses by SARS-CoV-2. Nature Communications 11: 3810.
Mulchandani, R., et al. 2021. Deciphering the COVID-19 cytokine storm: systematic review and meta-analysis. European Journal of Clinical Investigation 51: e13429.
de la Rica, R., et al. 2020. COVID-19: in the eye of the cytokine storm. Front Immunology 11: 558898.
Xiong, J., et al. 2020. Impact of COVID-19 pandemic on mental health in the general population: A systematic review. Journal of Affective Disorders 277: 55–64.
Wu, N.C., et al. 2020. An alternative binding mode of IGHV3–53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Reports 33:108274.
Yang, Q., et al. 2020. Inhibition of SARS-CoV-2 viral entry upon blocking N- and O-glycan elaboration. Elife 9: e61552.
Karki, R., et al. 2021. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184: 149-168.e17.
Chua, R.L., et al. 2020. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nature biotechnology 38: 970–979.
Liao, M., et al. 2020. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nature medicine 26: 842–844.
Mathew, D., et al. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369: eabc8511.
Schulte-Schrepping, J., et al. 2020. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182: 1419–1440.
Su, H., et al. 2020. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney international 98: 219–227.
Wilk, A.J., et al. 2020. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nature medicine 26: 1070–1076.
Schultze, A., et al. 2020. Risk of COVID-19-related death among patients with chronic obstructive pulmonary disease or asthma prescribed inhaled corticosteroids: An observational cohort study using the OpenSAFELY platform. The Lancet Respiratory Medicine 8: 1106–1120.
Kuri-Cervantes, L., et al. 2020. Comprehensive mapping of immune perturbations associated with severe COVID-19. Science immunology 5: eabd7114.
Giamarellos-Bourboulis, E.J., et al. 2020. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell host & microbe 27: 992–1000.
Swanson, K.V., et al. 2019. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nature Reviews Immunology 19: 477–489.
Ruland, J. 2014. Inflammasome: Putting the pieces together. Cell 156: 1127–1129.
Patton, L.M., et al. 1995. Interleukin-1 beta-induced neutrophil recruitment and acute lung injury in hamsters. Inflammation 19: 23–29.
Kolb, M., et al. 2001. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. The Journal of Clinical Investigation 107: 1529–1536.
Azkur, A.K., et al. 2020. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 75: 1564–1581.
Alosaimi, B., et al. 2020. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine 126: 154895.
Min, C.K., et al. 2016. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Science and Reports 6: 25359.
Meduri, G.U., et al. 1995. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107: 1062–1073.
Park, W.Y., et al. 2001. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 164: 1896–1903.
Beigel, J.H., et al. 2005. Avian influenza A (H5N1) infection in humans. New England Journal of Medicine 353: 1374–1385.
Tumpey, T.M., et al. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310: 77–80.
Kobasa, D., et al. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445: 319–323.
Fu, B., et al. 2020. Why tocilizumab could be an effective treatment for severe COVID-19? Journal of Translational Medicine 18: 164.
Fung, S.Y., et al. 2020. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg Microbes Infect 9: 558–570.
Channappanavar, R., et al. 2016. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host & Microbe 19: 181–193.
Nieto-Torres, J.L., et al. 2014. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathogens 10: e1004077.
Fu, Y., et al. 2020. Understanding SARS-CoV-2-mediated inflammatory responses: From mechanisms to potential therapeutic tools. Virol Sin. https://doi.org/10.1007/s12250-020-00207-4.
Kuriakose, T., et al. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Science Immunology 1: aag2045.
Rebsamen, M., et al. 2009. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Reports 10: 916–922.
Yabal, M., et al. 2014. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Reports 7: 1796–1808.
Allen, I.C., et al. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565.
Hornung, V., et al. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology 9: 847–856.
Mariathasan, S., et al. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232.
Petrilli, V., et al. 2007. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation 14: 1583–1589.
Nieto-Torres, J.L., et al. 2015. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485: 330–339.
Zhou, R., et al. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225.
Chen, C.C., et al. 2011. ORF8a of SARS-CoV forms an ion channel: Experiments and molecular dynamics simulations. Biochimica et Biophysica Acta 1808: 572–579.
Castaño-Rodriguez, C., et al. 2018. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio 9: e02325–17.
Shi, C.S., et al. 2019. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 5: 101.
Astuti, I. 2020. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes and Metabolic Syndrome 14: 407–412.
Zhao, C., and W. Zhao. 2020. NLRP3 inflammasome—a key player in antiviral responses. Frontiers in Immunology 11: 211.
de Castro-Jorge, L.A., et al. 2019. The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLoS Pathogens 15: e1007934.
Bertocchi, I., et al. 2020. The hidden role of NLRP3 inflammasome in obesity-related COVID-19 exacerbations: lessons for drug repurposing. British Journal of Pharmacology 177: 4921–4930. https://doi.org/10.1111/bph.15229. Epub 2020 Aug 26. PMID: 32776354; PMCID: PMC7436458.
Yap, J.K., et al. 2020. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. The Journal of Immunology 205: 307–312.
van den Berg, D.F., et al. 2020. Severe COVID-19: NLRP3 inflammasome dysregulated. Frontiers in Immunology 11: 1580.
Rodrigues, T.S., et al. 2020. Inflammasome activation in COVID-19 patients. medRxiv.
Wozniak, A.L., et al. 2010. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathogens 6: e1001087.
Farag, N., et al., 2020. Viroporins and inflammasomes: a key to understand virus-induced inflammation. The International Journal of Biochemistry & Cell Biology 122: 105738.
Issa, E., et al. 2020. SARS-CoV-2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems 5: e00266–20.
De Diego, M.L., et al. 2014. Inhibition of NF-κBmediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. Journal of Virology 88: 913–924.
Ratajczak, M.Z., et al. 2020. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 34: 1726–1729.
Ribeiro, D.E., et al. 2020. Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Molecular Psychiatry 1–16.
Rodrigues, T.S., et al. 2021. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. Journal of Experimental Medicine 218: e20201707.
Bok, K., et al. 2021. Accelerated COVID-19 vaccine development: milestones, lessons, and prospects. Immunity.
McLellan, J.S., et al. 2013. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342: 592–598.
Crank, M.C., et al. 2019. A proof of concept for structure-based vaccine design targeting RSV in humans. Science 365: 505–509.
Pallesen, J., et al. 2017. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proceedings of the National Academy of Sciences 114: E7348–E7357.
Marian, A.J. 2021. Current state of vaccine development and targeted therapies for COVID-19: impact of basic science discoveries. Cardiovascular Pathology 50: 107278.
Funding
MI received supports from Japanese Society for the Promotion of Science (JSPS/OF322, ID No. P19108), Tokyo, Japan, and D. S. Kothari Fellowship (F.4–2/2006 (BSR)/BL/18–19/0117), University of Grant Commission, Govt. of India. SNMNU is thankful to King Khalid University (Grant No. 339). SAM received support from King’s College London.
Author information
Authors and Affiliations
Contributions
MI designed and conceived this manuscript. MI, SAM, SNMNU, and UO wrote this manuscript. SP and KK reviewed and edited this review.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for Publication
All the authors have approved to publish this manuscript.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Islamuddin, M., Mustfa, S.A., Ullah, S.N.M.N. et al. Innate Immune Response and Inflammasome Activation During SARS-CoV-2 Infection. Inflammation 45, 1849–1863 (2022). https://doi.org/10.1007/s10753-022-01651-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-022-01651-y