
Abstract
MicroRNAs are small non-coding RNA regulatory molecules that play an important role in the development and function of immune cells. MicroRNA-26a (miR-26a) exhibits anti-inflammatory immune effects on immune cells. However, the exact mechanism by which miR-26a plays an anti-inflammatory role remains unclear. Here, we report that miR-26a reduces inflammatory response via inhibition of prostaglandin E2 (PGE2) production by targeting cyclooxygenase-2 (COX-2). We found that miR-26a was downregulated in vitro and in vivo. The miR-26a mimic significantly decreased COX-2 protein levels, further inhibiting pro-inflammatory cytokine production in LPS-stimulated macrophages. We predicted that miR-26a could potentially target COX-2 in LPS-stimulated macrophages. Computational algorithms showed that the 3′-UTR of COX-2 mRNA contains a binding site for miR-26a. This putative targeting relationship between miR-26a and COX-2 was further confirmed by a dual-reporter gene assay. The anti-inflammatory effects of the miR-26a mimic were diminished by PGE2 supplementation. Importantly, miR-26a mimics protected mice from lethal endotoxic shock and attenuated pro-inflammatory cytokine production. Collectively, these results suggest that miR-26a may function as a novel feedback negative regulator of the hyperinflammatory response and as a drug target for the progression of inflammation.
Similar content being viewed by others

INTRODUCTION
Inflammation is a “double-edged sword.” On the one hand, it can help the human body to resist pathogen infection and restore tissue structure and physiological function [1, 2]; on the other hand, uncontrolled inflammation can cause tissue damage and host diseases, such as septic shock [3], organ failure [4], cardiovascular diseases [5], and cancer [6]; even novel coronavirus (COVID-19) patients were reported to experience an inflammatory cytokine storm [7, 8]. Macrophages play a vital role in orchestrating the inflammatory response by producing a variety of inflammatory factors, including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1β [9, 10]. However, uncontrolled production of these cytokines causes hyperinflammation. Thus, it is necessary to identify a novel mechanism for the inhibition of the inflammatory reaction of macrophages.
MicroRNAs (miRNAs) are small non-coding RNAs (average length of 22 nucleotides) that regulate gene expression at the transcriptional and post-transcriptional levels [11, 12]. miRNAs regulate diverse biological processes, including the development, differentiation, and apoptosis of immune cells, morphogenesis, and pathological events [13,14,15]. Previous studies have reported that miR-146a, miR-let-7c, and miR-21 exhibit anti-inflammatory characteristics [16,17,18]. Overexpression of miR-146a significantly reduced IRAK1 expression, p65 levels in the nucleus, and IL-6 and TNF-α levels in the supernatant of THP-1 cell cultures. The overexpression of miR-let-7c in alveolar macrophages inhibited the expression of TNF-α, IL-6, and IL-1β by targeting STAT3. In a murine periodontitis model, maxillary molar ligation exacerbated gingival inflammation and alveolar bone loss in miR-21-deficient mice than their wild-type littermates. Recently, a study found that miR-26a was downregulated in hepatocellular carcinoma tissues. Importantly, this downregulation was associated with increased IL-6 expression [19]. Subsequently, miR-26a was found to markedly reduce IL-6, IL-1β, and TNF-α expression by targeting high-mobility group AT-hook 2 in lipopolysaccharide (LPS)-treated microglial cells [20]. Furthermore, miR-26a overexpression in mouse microglial cells significantly suppressed pro-inflammatory cytokine expression in mouse brains and markedly improved the neurological behavior and rotarod test performance of mice after intracerebral hemorrhage. Nevertheless, the suppressive role of miR-26a during the inflammatory response of macrophages remains largely unknown and requires further research.
Cyclooxygenase-2 (COX-2) is the rate-limiting enzyme in the metabolic conversion of arachidonic acid into various prostaglandins (PGs), including prostaglandin E2 (PGE2). Generally, COX-2 is undetectable in most normal tissues but is greatly induced in various cell types, including macrophages, in response to pro-inflammatory cytokines, mitogens, and tumor promoters [21]. PGE2 is a principal mediator of inflammation in diseases such as rheumatoid arthritis and osteoarthritis (OA). Nonsteroidal anti-inflammatory medications and selective COX-2 inhibitors reduce PGE2 production to diminish the inflammation observed in these diseases [22]. Additionally, miR-26a/-26b target COX-2 and regulate allergic inflammation-promoted tumorigenic and metastatic potential of cancer cells [23]. Downregulated miR-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2 [24]. Based on the concept that inflammation is a critical component of tumor progression, we hypothesized that miR-26a interacts with COX-2 to regulate inflammation.
In this study, we demonstrated that miR-26a reduced inflammation and cytokine production via PGE2 production by targeting COX-2. PGE2 supplementation significantly inhibited the anti-inflammatory effects of miR-26a. In addition, the miR-26a mimic relieved the death rate after endotoxin exposure in mice. Collectively, these findings reveal a novel anti-inflammatory modulation of miR-26a and provide guidance on the clinical use of miR-26a.
MATERIALS AND METHODS
Mice
C57BL/6J mice were from the Lab Animal Center of Southern Medicine University (Guangzhou, China). All mice were maintained under specific pathogen-free conditions in the Lab Animal Center of Southern Medicine University. All animal experiments in this study were approved by the Medical Ethics Board and the Biosafety Management Committee of Southern Medical University.
Mouse Peritoneal Macrophages Isolation
Macrophages were obtained from the peritoneal cavity of 6–8 weeks old C57BL/6J mice that were treated with 3% sterile thioglycolate 4 days before. Harvested cells were cultured at a density of 1 × 106 cells/well in 24-well flat-bottom culture plates in RPMI medium supplemented with 10% FBS, 100 IU/mL penicillin, and 100 mg/mL streptomycin. The next day non-adherent cells were removed by vigorous washing.
Mouse Monocytes Isolation
Leukocyte pools from C57BL/6J male mice 6–8 weeks of age, bled by cardiac puncture, were subjected to red blood cell lysis. Cells were harvested, washed, and stained for 30 min on ice with mixtures of fluorescently conjugated mAbs or isotype-matched controls. Mouse mAbs were PE-anti-CD11b (clone: M1/70; eBioscience, USA) and FITC-anti-Gr-1 (clone: RB6-8C5; eBioscience, USA). Thereafter, monocyte subsets (CD11b+ GR-1−) were sorted with a FACS Aria (BD Biosciences).
In Vivo Experiments
C57BL/6J mice were intraperitoneally (i.p.) injected with LPS (5 mg/kg, from E. coli 0111:B4, Sigma-Aldrich, USA) with or without miR-26a mimic (Ribo Bio, China). For miR-26a mimic-treated mice, the mimics dose (100 μL/mouse) was equivalent to 10 mg miR-26a/kg body weight on day one according to a previous report [25]. The miR-26a mimic and LPS were dissolved in 0.9% normal saline for in vivo experiments. Mice in the control group were injected with saline of the same volume with the same method. Solutions were prepared fresh immediately before injection. Serum samples were collected 24 h later. In detail, the blood was taken from the mouse eye socket without any anticoagulant and transferred to sterile tubes which were then incubated in a standing position, for 30 min. Thereafter, samples were centrifuged (1500 g, 10 min, at 4 °C) to recover the top fraction (serum) to be analyzed in ELISA. The lethal endotoxic shock was induced in C57BL/6J mice by i.p. LPS injection (10 mg/kg) according to a previous report [25]. The death of mice was judged according to whether the mice had a heartbeat or not.
Cell Culture and Treatment
Bone marrow cells were taken from C57BL/6J mice and placed on cell culture dishes (96 mm × 22 mm; CELLTER, China) at 37 °C/5% CO2 in DMEM (Corning, USA) containing 10% fetal bovine serum (FBS; Corning, USA). The cells differentiated into macrophages induced by granulocyte macrophage colony-stimulating factor (GM-CSF, 100 ng/mL; PeproTech, USA) for 7 days. BMDMs were placed on 12-well cell culture plates (CELLTER) for 48 h at 37 °C/5% CO2 in DMEM containing 10% FBS. Then, mouse macrophages were cultured with LPS (from E. coli K235, 1 μg/mL for all in vitro stimulations) for the specified time.
Human monocytic cell line THP-1 was purchased from Shanghai Cell Bank, Chinese Academy of Sciences. The cells were cultured in RPMI-1640 (Hyclone, Thermo Scientific, USA) medium supplemented with 10% fetal bovine serum (FBS; Corning, USA) at 37 °C in a humidified incubator with 5% CO2 and 95% air atmosphere. To stimulate differentiation, cells (5 × 105 cells/mL) were cultured with 100 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, USA) for 24 h. Then non-attached cells were removed by aspiration and the adherent cells were washed with RPMI 1640 for three times. For cell stimulation, the adherent cells followed by 24-h incubation were further incubated with or without lipopolysaccharide for the specified time.
RNA Interference
miRNA-26a mimics, miRNA mimics negative control (mimic NC), miR-26a inhibitor, miRNA inhibitor negative control (inhibitor NC), and COX-2-siRNA were synthesized and purified by Guangzhou Ribo Bio Co. Transfection was performed with ribo FECT™CP Transfection Kit (RN: R10035.3, RiboBio, China). In detail, macrophages were seeded into 6-well plates 1 day prior to transfection. And then, mimic (30 nM) or inhibitor (50 nM) or COX-2 siRNA (100 nM) and its negative control was transfected into the cells according to the manufacturer’s instructions.
PGE2 Supplementation Test
BMDMs were transfected with control or miR-26a mimic together with or without PGE2 addition for 24 h followed by incubated with LPS for an additional 24 h. TNF-α, IL-6, and IL-1β concentrations in culture supernatants were measured by ELISA.
Quantitative PCR Analysis
The experiments were performed as previously described [26]. Total RNA was isolated with TRIzol Reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. For the detection of mature miRNAs, small RNA levels were analyzed by real-time PCR using a SYBR® PrimeScript™ miRNA RT-PCR Kit (TaKaRa, Bio, Japan) in the method of poly-A. The expression of miR-26a was normalized against U6 snRNA levels, using the forward primer 5′-CGTTCAAGTAATCCAGGATAGGC-3′ and the universal reverse primer (TaKaRa, Bio, Japan). For the detection of mRNA, total RNA was reverse transcribed to cDNA primed by Prime ScriptTMRT reagent Kit (TaKaRa, Bio, Japan) and subsequently by PCR using SYBR Premix ExTaq™ (TaKaRa, Bio, Japan), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control gene: COX-2 forward 5′-GGCGGCTCGAGAAAGGAGTCATACTTGTGAA-3′, reverse 5′-AATGCGGCCGC GACAGCTTCTTTTTGGTATA-3′, GAPDH forward 5′-TCGACAGT CAGCCGCATCTTCTTT-3′, reverse 5′-GCCCAATACGACCAAATCCGTTGA-3′.
Bioinformatics Method, 3′-UTR Datasets, and microRNA Datasets
MiRNA target site prediction for COX-2 was performed using Target scan Release 4.0 (http://www.targetscan.org). The 3′-UTR sequences of COX-2 were retrieved using Ensembl Data base (http://www.ensembl.org). Human miRNA sequences of the miR-26a registry were downloaded from the miRBase website (http://www.mirbase.org).
Vector Construction
To construct the luciferase report vector, COX-2 3′-UTR and its flanking sequence was PCR-amplified using primers forward 5′-GGCGGCTCGAGAAAGGAGTCATACTTGTGAA-3′ and reverse 5′-AATGCGGCCGCGACAGCTTCTTTTTGGTATA-3′. Similarly, a mutated COX-2 3′-UTR fragment in which the miR-26a binding site was mutated was PCR-amplified using primers forward 5′-GACGTCTTTAATGAACTTTTTCAACTTATATTATAAG-3′ and reverse 5′-AAGTTGAAAAAGTTCATTAAAGACGTCAAAACTCATT-3′. The PCR product was cloned into vector pmiR-RB-REPORT™ (Ribo. Co, China) that vector reporter fluorescence is hRluc, while luc as a control for normalization.
Dual-Luciferase Activity Assay
The 293 cells were seeded in 96 wells and transfected using Lipofectamine™ 2000 (Promega, USA) with pmiR-RB-REPORT™/COX-2 3′-UTR (500 ng), pmiR-RB-REPORT™/C COX-2 3′-UTR mutant (500 ng), miR-26a mimics (50 nM), or miR-NC (50 nM). The cell lysate was collected and assayed 48 h after transfection. Luciferase activities of WT-luc and Mut-luc were measured using a Dual-Luciferase Reporter Assay System (Promega, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
TNF-α, IL-6, and IL-1β levels in culture supernatant and mouse serum were measured by enzyme-linked immunosorbent assay kit (ExCell Bio, China) according to the manufacturer’s protocol. In detail, dilution factors were different when serum or culture supernatant tests were performed. Serum was diluted 1:1 (serum:diluent), and culture supernatant was diluted 1:2 (serum:diluent).
Western Blot Analysis
The experiments were performed as previously described [27, 28]. Total proteins were extracted from macrophages and quantified using the BCA protein assay kit (Beyotime, China). Total proteins (30 or 50 mg) from samples were separated by SDS-PAGE and blotted onto a PVDF membrane (Millipore Corp, Billerica, MA, USA). After blocking, nitrocellulose blots were incubated for 1 h with primary antibodies diluted in TBS/Tween 20 (0.075%) containing 3% dried skimmed milk powder (Marvel). Rabbit monoclonal antibody directed against COX-2 (clone: D5H5; CST # 12282) was used at 1:1000 as was anti-β-actin (Santa Cruz, USA, sc-2357). Horseradish peroxidase-conjugated anti-rabbit antibodies were used as secondary antibodies correspondingly. After washing, the membrane was processed using the enhanced chemiluminescence system. Proteins were visualized with an ECL-chemiluminescent kit (ECL-plus, Thermo Scientific, USA).
Statistics
All experiments were performed thrice in triplicate. When shown, multiple samples represent biological (not technical) replicates of mice randomly sorted into each experimental group. No blinding was performed during animal experiments. The data were analyzed for normality (one sample K-S test) and showed normal distribution. Determination of statistical differences was performed with Prism 5 (Graphpad Software, Inc.) using unpaired two-tailed t-tests (to compare two groups with similar variances) or one-way ANOVA with Bonferroni’s multiple comparison test (to compare more than two groups). Difference between mouse survival curves was evaluated by the log-rank (Mantel-Cox) test. P ≤ 0.05 was considered significant.
RESULTS
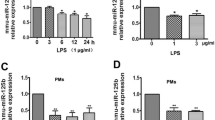
To measure the expression of miR-26a during the inflammatory process in macrophages in vitro, the human monocytic leukemia cell line THP-1 and murine bone marrow-derived macrophages (BMDMs) were used. First, THP-1 cells were induced to become macrophages (named THP-1 Mφ) by phorbol myristate acetate. We found that the expression of miR-26a was decreased in THP-1 Mφ cells after LPS stimulation (Fig. 1A). Similarly, miR-26a expression was reduced in BMDMs treated with LPS (Fig. 1B). To further determine the expression pattern of miR-26a during the inflammatory process in vivo, we challenged mice with LPS intraperitoneally and tested miR-26a expression in peritoneal exudate macrophages. The results showed that the expression of miR-26a was downregulated compared to that of monocytes from peripheral blood (Fig. 1C). Together, these results suggest that miR-26a is downregulated in macrophages during inflammation.

miR-26a expression is reduced in macrophages after LPS stimulation. A THP-1 cells were stimulated with PMA for 24 h and then stimulated with LPS (100 ng/mL) for the indicated times. The expression levels of miR-26a were measured by real-time PCR. B Murine bone marrow-derived macrophages (BMDMs) were stimulated with LPS (100 ng/mL) for the indicated times, and the expression levels of miR-26a were measured by real-time PCR. C The mice were intraperitoneally challenged the mice with LPS (5 mg/kg body weight) for 48 h, and peritoneal exudate macrophage were isolated. The expression levels of miR-26a were examined by real-time PCR. Monocytes isolated from peripheral blood were as control. Data shown in are the mean ± SD. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Data are representative of three independent experiments with similar results. Values in (A and B) were compared using one-way ANOVA with Bonferroni’s test, and values in C were compared using t test.
MiR-26a Inhibits Pro-inflammatory Cytokine Production in Macrophages
To explore the role of miR-26a in inflammatory immune responses, macrophages were transfected with a chemically synthesized miR-26a mimic. Real-time polymerase chain reaction showed that miR-26a levels increased dramatically after mimic transfection, confirming the efficacy of overexpression (Fig. 2A). The production of TNF-α, IL-6, and IL-1β was decreased in BMDMs with the miR-26a mimic after LPS stimulation (Fig. 2B). Moreover, an inhibitor was used to antagonize endogenous miR-26a to validate its role in inflammatory immune responses. The level of miR-26a was dramatically reduced when a specific inhibitor was transfected into BMDMs (Fig. 2C). Consistent with the above results, inhibition of miR-26a enhanced TNF-α, IL-6, and IL-1β secretion (Fig. 2C). We then validated these results in THP-1 Mφ cells following LPS stimulation. Similarly, we found that miR-26a mimics reduced TNF-α, IL-6, and IL-1β production (Supplementary Fig. 1A, B), but the addition of the miR-26a inhibitor led to the opposite outcome (Supplementary Fig. 1C, D). Taken together, these results suggest that miR-26a inhibits pro-inflammatory cytokine production in macrophages.

miR-26a inhibits the production of inflammation cytokines in BMDMs. A BMDMs transfected with miR-26a mimics (50 nM) for 24 h, and then, qPCR analysis for the expression of miR-26a was performed. B BMDMs transfected with miR-26a mimics (50 nM) for 24 h and then incubated with LPS for the indicated times. The culture supernatants were collected. ELISA assay was used to test the concentrations of TNF-α, IL-6, and IL-1β in culture supernatants. C BMDMs were pre-treated with miR-26a inhibitors (100 nM) for 24 h, and then, qPCR analysis was performed to measure the expression of miR-26a. D BMDM were pre-treated with miR-26a inhibitors (100 nM) for 24 h and then incubated with LPS for the indicated times. The culture supernatants were collected. ELISA assay was used to test the concentrations of TNF-α, IL-6, and IL-1β in culture supernatants. Data shown are the mean ± SD. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Data are representative of three independent experiments with similar results. Values in A and C were compared using t-test and using two-way ANOVA with Bonferroni’s test in B and D.
MiR-26a Represses COX-2 Expression Post-transcriptionally to Inhibit Secretion of Pro-inflammatory Cytokines
To identify the specific target of miR-26a that modulates pro-inflammatory cytokine production, bioinformatics analysis was performed using TargetScan (http://www.targetscan.org). We found that COX-2 displayed a potential evolutionarily conserved seed match for miR-26a in its 3′-UTR (Fig. 3A). Next, sequences from the miR-26a target site in the 3′-UTR of COX-2 and its mutant variants were successfully cloned into pmiR-RB-REPORT™ vectors. The vectors containing the 3′-UTR of COX-2 (WT-Cox-2) and its mutant variants (Mut-Cox-2) were transfected into 293 cells. In luciferase reporter assays, transfection with miR-26a mimics significantly inhibited the chemiluminescence of 293 cells transfected with WT-Cox-2 compared to the negative controls (Fig. 3B). However, miR-26a mimics had no effect on the chemiluminescence of 293 cells transfected with Mut-Cox-2 fragments in which the nine bases in the miR-26a target site were mutated. Our results indicate that these sequences from the 3′-UTR of mouse COX-2 are sufficient for miR-26a binding and regulation. To confirm whether miR-26a targets COX-2, we examined COX-2 expression in BMDMs or THP-1 Mφ transfected with control or miR-26a mimic. As expected, miR-26a did not affect the mRNA level of COX-2 (Fig. 3C and Supplementary Fig. 2A) but substantially reduced COX-2 protein levels (Fig. 3D and Supplementary Fig. 2B). Moreover, we used an miR-26a inhibitor to further confirm whether miR-26a targeted COX-2 in BMDMs or THP-1 Mφ. Similarly, COX-2 was not influenced by the miR-26a inhibitor at the mRNA level (Fig. 3E and Supplementary Fig. 2C) but rather at the protein level (Fig. 3F and Supplementary Fig. 2D).

miR-26a represses COX-2 expression post-transcriptionally to inhibit the secretion of pro-inflammatory cytokines. A Sequence of miR-26a and its predicted binding sites within the COX-2 3-′UTRs are shown. B 293 cells were co-transfected with control or miR-26a mimic and a wild-type (WT-COX-2) or mutated 3-′UTR (mut-COX-2) luciferase reporter plasmid and assessed for luciferase activity at 48 h after transfection. C–D BMDMs were transfected with control or miR-26a mimics (50 nM) for 24 h and then incubated with LPS for 24 h. The mRNA (C) and protein (D) levels of COX-2 were measured by real-time PCR and Western blot, respectively. (E–F BMDMs were transfected with control or miR-26a inhibitor (100 nM) for 24 h and then incubated with LPS for 24 h. The mRNA (E) and protein (F) levels of COX-2 were measured by real-time PCR and Western blot, respectively. G–H BMDMs were transfected with negative control siRNA (siNC) or COX-2 siRNA, followed by LPS infection for 24 h. G The mRNA levels of COX-2 were examined. H TNF-α, IL-6, and IL-1β concentrations in culture supernatants were measured by ELISA respectively. Data shown are the mean ± SD. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Data are representative of three independent experiments with similar results. Values in G were compared using t-test and values in B–F, and H was compared using two-way ANOVA with Bonferroni’s test.
To confirm whether miR-26a regulated LPS-triggered cytokine production by targeting COX-2 in macrophages, BMDMs and THP-1 Mφ were transfected with COX-2 siRNA to knock down the expression of COX-2. The endogenous expression of COX-2 mRNA was significantly downregulated with COX-2 siRNA in BMDMs and THP-1 Mφ (Fig. 3G and Supplementary Fig. 2E). The knockdown of COX-2 markedly reversed the LPS-induced release of TNF-α, IL-6, and IL-1β (Fig. 3H and Supplementary Fig. 2F). These results suggest that miR-26a reduces the production of TNF-α, IL-6, and IL-1β by targeting COX-2.
Anti-inflammatory Effect of miR-26a Depends on PGE2 Production in Macrophages
PGE2 is the major metabolic product of COX-2, critical for the progression of pain, and inflammation. We hypothesized that miR-26a regulates LPS-triggered cytokine production via PGE2 in macrophages. To confirm this hypothesis, BMDMs or THP-1 Mφ were pre-transfected with a miR-26a mimic or inhibitor. We found that the miR-26a mimic reduced PGE2 production after LPS stimulation (Fig. 4A and Supplementary Fig. 3A) and that the miR-26a inhibitor enhanced PGE2 production (Fig. 4B and Supplementary Fig. 3B). Moreover, the reduced expression of TNF-α, IL-6, and IL-1β regulated by miR-26a mimic was abolished by PGE2 supplementation in BMDMs (Fig. 4C) or THP-1 Mφ (Supplementary Fig. 3C) following LPS stimulation. Overall, these results indicate that miR-26a plays an anti-inflammatory role by reducing the secretion of PGE2 downstream of COX-2.

miR-26a regulates pro-inflammatory cytokines via PGE2 production. BMDMs were pre-treated with miR-26a mimic (50 nM) (A) or miR-26a inhibitors (100 nM) (B) for 24 h and incubated with LPS for the specified time. PGE2 concentrations in culture supernatants were measured by ELISA. C BMDMs were transfected with control or miR-26a mimic together with or without PGE2 addition for 24 h followed by incubated with LPS for 24 h. TNF-α, IL-6, and IL-1β concentrations in culture supernatants were measured by ELISA respectively. Data shown in are the mean ± SD. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Data are representative of three independent experiments with similar results. Values in A–B were compared using two-way ANOVA with Bonferroni’s test, and values in C were compared using one-way ANOVA with Bonferroni’s test.
MiR-26a Reduces Inflammatory Responses In Vivo
In vitro assays showed that miR-26a suppressed the inflammatory response by reducing PGE2 secretion. To further confirm this mechanism, in vivo animal experiments were performed. A hyperinflammatory response can lead to pathological damage and death. To test the anti-inflammatory effect of miR-26a, a high dose of LPS was intraperitoneally injected into mice to induce lethal endotoxic shock. The initial time of death was delayed, and the survival rate was improved in 3 treated with the miR-26a mimic compared to control mice (Fig. 5A). COX-2 expression was significantly reduced in peritoneal exudate macrophages from mice treated with the miR-26a mimic compared to those from control mice (Fig. 5B). Mice pre-treated with the miR-26a mimic had lower serum PGE2 production than control mice (Fig. 5C), as well as lower levels of TNF-α, IL-6, and IL-1β in serum (Fig. 5D). Taken together, these results suggest that miR-26a reduces inflammatory responses via PGE2 production by targeting COX-2 in vivo.

miR-26a reduces LPS-induced inflammatory response in vivo. C57BL/6J mice were tail vein injected with control solution or miR-26a mimic (10 mg/kg bodyweight) and then intraperitoneally injected with saline or LPS (10 mg/kg bodyweight) 24 h later (n = 10 mice for each group) on day one. A The survival rate of the mice was assessed. B After 48 h, peritoneal exudate macrophage were isolated (n = 5 mice for each group). The expression levels of COX-2 were examined by Western blot. C–D After 48 h, serum samples were collected (n = 5 mice for each group). C The PGE2 concentration was quantified by ELISA. D TNF-α, IL-6, and IL-1β concentrations were quantified by ELISA. Data shown in are the mean ± SD. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Data are representative of three independent experiments with similar results. Values in B and C were compared using one-way ANOVA with Bonferroni’s test.
DISCUSSION
Macrophages activated by LPS produce a series of pro-inflammatory mediators to detect the presence of pathogens and help trigger the immune response [29, 30]. The immune regulatory effects of miRNAs on macrophages have drawn great attention. The immune modulation of various miRNAs, such as miR-146a, let7c, and miR-155, has been investigated [31,32,33]. Here, we provide evidence related to the anti-inflammatory activity of miR-26a in LPS-induced acute infection and septic shock via PGE2 production.
Previous reports have shown that miR-26a acts as an important inflammatory regulator in several diseases. Patients with relapsing–remitting multiple sclerosis, which is one of the most common idiopathic inflammatory demyelinating diseases of the central nervous system, had a higher miR-26a-5p expression in the blood after 3 months of IFN-β treatment [34]. In the synovial tissues of OA rats, miR-26a mimics reduced the protein levels of p-IκBα and p-P65. Thus, synovial tissue hyperplasia, inflammation, and cartilage injury of the femoral condyle were ameliorated, with decreased inflammation and cartilage injury scores and synoviocyte and chondrocyte apoptosis [35]. There is a positive correlation between the expression of miR-26a, miR-146a, and miR-31 and the production of IL-5, IL-8, IL-12, and TNF-α [36]. A recent study showed that miR-26a overexpression markedly alleviated chemically induced and palmitate-triggered ER stress and lipid accumulation in human hepatoma cells and murine primary hepatocytes [37]. In addition, a lower baseline miR-26a expression in PBMCs indicated the occurrence of early-onset neonatal sepsis, and a reduced miR-26a expression might be partly related to the inflammatory process [38]. In our study, we showed that the expression levels of miR-26a were downregulated following LPS stimulation, indicating that miR-26a may be involved in macrophage regulation. Furthermore, overexpression of miR-26a was associated with a significant decrease in the production of TNF-α, IL-6, and IL-1β, while knockdown of miR-26a exhibited a reverse effect in regulating the production of these proteins, consistent with previous research [20, 39]. However, it is regrettable that there is a relatively modest effect in survival outcomes from mice with LPS-induced excessive inflammation after miR-26a administration. How to improve the anti-inflammatory ability of miR-26a in vivo needs further study.
The role of PGE2 in inflammation is ambiguous. PGE2 induced by LPS generates deleterious effects in neurons, leading to lesions or faster pain transmission [40,41,42]. However, PGE2 also has anti-inflammatory properties. It participates in the neuroprotection induced by bradykinin and blocks cytokine synthesis activated by LPS or ATP in microglia [43, 44]. Our data confirmed that PGE2 was sufficient to induce LPS-induced inflammation. However, no blinding was performed during the experiments. Further experiments are required to confirm these results.
The mechanisms by which miRNAs induce significant degradation of mRNA targets and repress protein synthesis remain poorly understood. In our study, we confirmed that the 3′-UTR of COX-2 mRNA contained a binding site for miR-26a, and the modifications of miR-26a function exhibited corresponding repression or augmentation of COX-2 expression at the protein but not the mRNA level. One possible explanation might be that miR-26a post-transcriptionally controls COX-2 protein expression, which was supported by Kwon et al. [23], Zhang et al. [24], and Jin et al. [45]. The underlying mechanisms require further investigation. In addition, our findings reveal not only the regulation of miR-26a in COX-2 expression, but also cytokine production in LPS-stimulated macrophages.
In conclusion, our study identified the reduction of miR-26a and induction of COX-2 in LPS-stimulated macrophages and revealed that miR-26a-mimic administration led to the suppression of COX-2 protein expression and LPS-induced cytokine production. These findings demonstrate that miR-26a inhibits COX-2 expression, implicating miR-26a as a drug target for the progression of inflammation.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
References
Gong, T., L. Liu, W. Jiang, and R. Zhou. 2020. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nature Reviews Immunology 20: 95–112.
Medzhitov, R. 2010. Inflammation 2010: New adventures of an old flame. Cell 140: 771–776.
Korneev, K.V. 2019. Mouse models of sepsis and septic shock. Molekuliarnaia Biologiia (Mosk) 53: 799–814.
Fowler, A.A., 3rd., J.D. Truwit, R.D. Hite, P.E. Morris, and C. DeWilde. 2019. Effect of vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: The CITRIS-ALI randomized clinical trial. JAMA 322: 1261–1270.
Taube, A., R. Schlich, H. Sell, K. Eckardt, and J. Eckel. 2012. Inflammation and metabolic dysfunction: Links to cardiovascular diseases. American Journal of Physiology. Heart and Circulatory Physiology 302: H2148-2165.
Greten, F.R., and S.I. Grivennikov. 2019. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 51: 27–41.
Mahmudpour, M., J. Roozbeh, M. Keshavarz, S. Farrokhi, and I. Nabipour. 2020. COVID-19 cytokine storm: the anger of inflammation. Cytokine 133: 155151.
McGonagle, D., K. Sharif, A. O’Regan, and C. Bridgewood. 2020. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev 19: 102537.
Murray, P.J. 2017. Macrophage polarization. Annual Review of Physiology 79: 541–566.
Arora, S., K. Dev, B. Agarwal, P. Das, and M.A. Syed. 2018. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 223: 383–396.
Bartel, D.P. 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233.
Fabian, M.R., N. Sonenberg, and W. Filipowicz. 2010. Regulation of mRNA translation and stability by microRNAs. Annual Review of Biochemistry 79: 351–379.
Kiselev, F.L., M. Li, J. Li, X. Ding, M. He, and S.Y. Cheng. 2014. [MicroRNA and cancer] microRNA and cancer. Molekuliarnaia Biologiia (Mosk) 48: 232–242.
Gross, N., J. Kropp, and H. Khatib. 2017. MicroRNA signaling in embryo development. Biology (Basel) 6.
Lee, H.M., D.T. Nguyen, and L.F. Lu. 2014. Progress and challenge of microRNA research in immunity. Frontiers in Genetics 5: 178.
Yu, J.H., L. Long, Z.X. Luo, L.M. Li, and J.R. You. 2016. Anti-inflammatory role of microRNA let-7c in LPS treated alveolar macrophages by targeting STAT3. Asian Pacific Journal of Tropical Medicine 9: 72–75.
Zhou, W., L. Su, X. Duan, X. Chen, A. Hays, S. Upadhyayula, J. Shivde, H. Wang, Y. Li, D. Huang, and S. Liang. 2018. MicroRNA-21 down-regulates inflammation and inhibits periodontitis. Molecular Immunology 101: 608–614.
Zhou, C., L. Zhao, K. Wang, Q. Qi, M. Wang, L. Yang, P. Sun, and H. Mu. 2019. MicroRNA-146a inhibits NF-κB activation and pro-inflammatory cytokine production by regulating IRAK1 expression in THP-1 cells. Experimental and Therapeutic Medicine 18: 3078–3084.
Yang, X., L. Liang, X.F. Zhang, H.L. Jia, Y. Qin, X.C. Zhu, X.M. Gao, P. Qiao, Y. Zheng, Y.Y. Sheng, J.W. Wei, H.J. Zhou, N. Ren, Q.H. Ye, Q.Z. Dong, and L.X. Qin. 2013. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology 58: 158–170.
Jin, J., F. Zhou, J. Zhu, W. Zeng, and Y. Liu. 2020. MiR-26a inhibits the inflammatory response of microglia by targeting HMGA2 in intracerebral hemorrhage. Journal of International Medical Research 48: 300060520929615.
Patrono, C. 2016. Cardiovascular effects of cyclooxygenase-2 inhibitors: A mechanistic and clinical perspective. British Journal of Clinical Pharmacology 82: 957–964.
Park, J.Y., M.H. Pillinger, and S.B. Abramson. 2006. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clinical Immunology 119: 229–240.
Kwon, Y., Y. Kim, S. Eom, M. Kim, D. Park, H. Kim, K. Noh, H. Lee, Y.S. Lee, J. Choe, Y.M. Kim, and D. Jeoung. 2015. MicroRNA-26a/-26b-COX-2-MIP-2 loop regulates allergic inflammation and allergic inflammation-promoted enhanced tumorigenic and metastatic potential of cancer cells. Journal of Biological Chemistry 290: 14245–14266.
Zhang, J., J. Liang, and J. Huang. 2016. Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncology Letters 12: 3397–3402.
Du, X., Y. Yang, X. Zhan, Y. Huang, Y. Fu, Z. Zhang, H. Liu, L. Zhang, Y. Li, Q. Wen, X. Zhou, D. Zuo, C. Zhou, L. Li, S. Hu, and L. Ma. 2020. Vitamin B6 prevents excessive inflammation by reducing accumulation of sphingosine-1-phosphate in a sphingosine-1-phosphate lyase-dependent manner. Journal of Cellular and Molecular Medicine 24: 13129–13138.
Hu, S., X. Du, Y. Huang, Y. Fu, Y. Yang, X. Zhan, W. He, Q. Wen, X. Zhou, C. Zhou, X.P. Zhong, J. Yang, W. Xiong, R. Wang, Y. Gao, and L. Ma. 2018. NLRC3 negatively regulates CD4+ T cells and impacts protective immunity during Mycobacterium tuberculosis infection. PLoS pathogens 14: e1007266.
Fu, Y., P. Wang, J. Zhao, Y. Tan, J. Sheng, S. He, X. Du, Y. Huang, Y. Yang, J. Li, Y. Cai, Y. Liu, and S. Hu. 2021. USP12 promotes CD4(+) T cell responses through deubiquitinating and stabilizing BCL10. Cell Death & Differentiation.
Fu, Y., X. Zhan, Y. Wang, X. Jiang, M. Liu, Y. Yang, Y. Huang, X. Du, X.P. Zhong, L. Li, L. Ma, and S. Hu. 2019. NLRC3 expression in dendritic cells attenuates CD4(+) T cell response and autoimmunity. EMBO Journal 38: e101397.
Wang, S., D. Xiang, F. Tian, and M. Ni. 2021. Lipopolysaccharide from biofilm-forming Pseudomonas aeruginosa PAO1 induces macrophage hyperinflammatory responses. Journal of Medical Microbiology 70.
Lin, C.Y., S.H. Kao, L.C. Hung, H.J. Chien, W.H. Wang, Y.W. Chang, and Y.H. Chen. 2021. Lipopolysaccharide-induced nitric oxide and prostaglandin E2 production is inhibited by tellimagrandin II in mouse and human macrophages. Life (Basel) 11.
Wang, H., X. Li, T. Li, L. Wang, X. Wu, J. Liu, Y. Xu, and W. Wei. 2019. Multiple roles of microRNA-146a in immune responses and hepatocellular carcinoma. Oncology Letters 18: 5033–5042.
Mahesh, G., and R. Biswas. 2019. MicroRNA-155: A master regulator of inflammation. Journal of Interferon and Cytokine Research 39: 321–330.
Essandoh, K., Y. Li, J. Huo, and G.C. Fan. 2016. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 46: 122–131.
Havelange, V., N. Stauffer, C.C. Heaphy, S. Volinia, M. Andreeff, G. Marcucci, C.M. Croce, and R. Garzon. 2011. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer 117: 4696–4706.
Zhao, Z., X.S. Dai, Z.Y. Wang, Z.Q. Bao, and J.Z. Guan. 2019. MicroRNA-26a reduces synovial inflammation and cartilage injury in osteoarthritis of knee joints through impairing the NF-κB signaling pathway. Bioscience Reports 39.
Shi, Z.G., Y. Sun, K.S. Wang, J.D. Jia, J. Yang, and Y.N. Li. 2019. Effects of miR-26a/miR-146a/miR-31 on airway inflammation of asthma mice and asthma children. European Review for Medical and Pharmacological Sciences 23: 5432–5440.
Xu, H., Y. Tian, D. Tang, S. Zou, G. Liu, J. Song, G. Zhang, X. Du, W. Huang, B. He, W. Lin, L. Jin, W. Huang, J. Yang, and X. Fu. 2021. An endoplasmic reticulum stress-MicroRNA-26a feedback circuit in NAFLD. Hepatology 73: 1327–1345.
Zhao, Z., and J. Zhang. 2021. Lower expression of miR-26a in PBMCs indicates the occurrence of early-onset neonatal sepsis and is partly mediated by the upregulation of PTEN. Frontiers in Pediatrics 9: 678205.
Kumar, A., H.S. Bhatia, A.C. de Oliveira, and B.L. Fiebich. 2015. microRNA-26a modulates inflammatory response induced by toll-like receptor 4 stimulation in microglia. Journal of Neurochemistry 135: 1189–1202.
Montine, T.J., D. Milatovic, R.C. Gupta, T. Valyi-Nagy, J.D. Morrow, and R.M. Breyer. 2002. Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. Journal of Neurochemistry 83: 463–470.
Reinold, H., S. Ahmadi, U.B. Depner, B. Layh, C. Heindl, M. Hamza, A. Pahl, K. Brune, S. Narumiya, U. Müller, and H.U. Zeilhofer. 2005. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. The Journal of Clinical Investigation 115: 673–679.
Hsieh, C.T., Y.J. Lee, J.W. Lee, S. Lu, M.A. Tucci, X. Dai, N.B. Ojeda, H.J. Lee, L.W. Fan, and L.T. Tien. 2020. Interleukin-1 receptor antagonist ameliorates the pain hypersensitivity, spinal inflammation and oxidative stress induced by systemic lipopolysaccharide in neonatal rats. Neurochemical International 135: 104686.
Caggiano, A.O., and R.P. Kraig. 1998. Prostaglandin E2 and 4-aminopyridine prevent the lipopolysaccharide-induced outwardly rectifying potassium current and interleukin-1beta production in cultured rat microglia. Journal of Neurochemistry 70: 2357–2368.
Noda, M., Y. Kariura, U. Pannasch, K. Nishikawa, L. Wang, T. Seike, M. Ifuku, Y. Kosai, B. Wang, C. Nolte, S. Aoki, H. Kettenmann, and K. Wada. 2007. Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. Journal of Neurochemistry 101: 397–410.
Jin, Z., J. Ren, and S. Qi. 2020. Human bone mesenchymal stem cells-derived exosomes overexpressing microRNA-26a-5p alleviate osteoarthritis via down-regulation of PTGS2. International Immunopharmacology 78: 105946.
Acknowledgements
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper.
Funding
This work was funded by grants from the National Natural Science Foundation of China (81901614, 32070906, and 81971805), Guangdong Basic and Applied Basic Research Foundation (2019A1515011103, 2020A1515010158).
Author information
Authors and Affiliations
Contributions
T.Y. and S.F.H. designed research; T.Y., P.W., Y.X.W., J.B.Z., Q.S.C., D.M.W., and H.C. conducted research; T.Y., P.W., S.F.H., and Q.S.W. analyzed data; T.Y. wrote the paper; P.W. and S.F.H. provided essential reagents or provided essential materials; S.F.H. and Q.S.W. as the corresponding author conducted the experiment. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
All animal experiments in this study were approved by the Medical Ethics Board and the Biosafety Management Committee of Southern Medical University.
Consent for Publication
I understand that my manuscript and associated personal data will be shared with Research Square for the delivery of the author dashboard.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, T., Wang, P., Wu, Y. et al. MiR-26a Reduces Inflammatory Responses via Inhibition of PGE2 Production by Targeting COX-2. Inflammation 45, 1484–1495 (2022). https://doi.org/10.1007/s10753-022-01631-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-022-01631-2