
Abstract
The Western Balkans hosts the richest subterranean aquatic gastropod fauna in the world. The main factors shaping intraspecies diversity are thought to be isolation and endemism. In the genus Belgrandiella, minute snails inhabiting subterranean waters and springs in Central Europe and Balkans, molecular studies have shown much fewer valid species than previously anticipated. The present study applies mitochondrial cytochrome c oxidase subunit I, histone 3, and RAPD analysis, to check the inter- and intraspecies genetic diversity in 36 Belgrandiella populations from caves, springs and interstitial aquifers. The level of gene flow is assessed to check if these snails form a widespread genetically uniform metapopulation or rather follow the highly endemic pattern. The studied populations have been assigned to six species. In the most widely distributed B. kusceri from 21 populations, 60 sequenced specimens represent 16 haplotypes. While the same haplotypes are present in distant populations, gene flow between the other populations is low. Nei distances for RAPD show no geographic pattern. The interspecies differences in COI evidently confirm the time of speciation in Pleistocene, before karstification, which rejects speciation within isolated caves. The pattern observed in Belgrandiella seems more similar to the one described in Montenegrospeum than in Kerkia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Freshwater gastropods are a noteworthy element of the subterranean fauna. According to Illies (1978), from among 549 freshwater gastropod species of Europe as many as 106 inhabit subterranean waters, nearly all as stygobites. Stygobites are obligate, strictly subterranean aquatic animals which complete their entire life in the lightless environment. Only a few represent either stygophiles, i.e., they inhabit both surface and subterranean aquatic environments, but are not necessarily restricted to either, or stygoxenes, i.e., they are accidentally or occasionally present in subterranean waters. In the Balkans, the gastropod stygofauna is especially rich, comprising 169 species (Sket et al., 2004a). Even the representatives of two endemic families (Emmericiidae, Litthabitellidae) could be found. Now we can count more than 300 nominal species, although the species distinctness of many of them needs verification (Falniowski, 2018; Osikowski et al., 2018). In Slovenia, from 91 aquatic gastropod species, 46 are stygobionts (50%), and eight either stygoxenes or stygophiles (Sket, 1999). Nearly all of the gastropod stygofauna represents the minute, gill breathing Truncatelloidea (“Hydrobiidae s. lato” in Bernasconi & Riedel, 1994; Culver, 2012). The Truncatelloidea are one of the greatest examples of the Balkans biodiversity (Bănărescu, 2004). Surely, such species numbers are preliminary estimates; on the one hand, several cryptic species may still be recognized since the subterranean habitats are underexplored, but on the other hand, the number of already recognized species is presumably overestimated (Falniowski & Beran, 2015; Osikowski et al., 2018). Minute dimensions and simplified anatomy of these snails (Falniowski, 2018) coupled with low densities of their populations (e.g., Culver & Pipan, 2009, 2014), result in poor knowledge of their biology, speciation, and taxonomy. Descriptions based usually only on the shell morphology resulted in numerous nominal species representing no real biological entities (e.g., Wilke & Falniowski, 2001; Osikowski et al., 2018). Long lasting tendency to describe a new species at each locality, based on the assumption that it is isolated, has resulted in e.g., more than 60 nominal species (and many subspecies) of Bythiospeum Bourguignat, 1882 listed by Glöer (2002), many of them not representing distinct species (Richling et al., 2016).
Despite common reports on the hydrobioid stygobiotic species endemic to extremely restricted area—often just one cave (for review see Falniowski et al., 2021a), the hydrobioid snail Fontigens tartarea Hubricht, 1963 is known from dozens of caves in West Virginia (North America). Its geographic range reaches 200 km, and the species shows patchy distribution, sporadic occurrence, and high variability of the shell (Hershler & Holsinger, 1990; Culver, 2012). Similarly, in Montenegrospeum bogici (Pešić et Glöer, 2012) inhabiting caves and interstitial waters in the Western Balkans, and sporadically flooded out in springs, a wide geographic range coupled with high levels of gene flow between local populations was found (Falniowski et al., 2021a). Haase et al. (2021) suggest possible dispersal and radiation along the alluvial gravel interstitial, as well as the phreatic rhizosphere (i.e., roots of macrophytes, either aquatic or hygrophilous plants) for Hauffenia Pollonera, 1898, a stygobiont snail inhabiting only interstitial waters, not caves in Slovakia. Although stygobites (e.g., Montenegrospeum mentioned above) tend to have wider geographic ranges than subterranean terrestrial troglobites (Lamoreaux, 2004). that is not the universal pattern. Several species of Kerkia Radoman, 1978, that inhabit subterranean waters in the same region of the Balkans, are highly geographically restricted. Above the species-level they form three clades, also reflecting geography, thus the level of endemism in Kerkia is high (Hofman et al., 2022a, b).
The genus Belgrandiella A. J. Wagner, 1928, inhabits subterranean waters and springs in Slovenia, Austria and northern Italy. Some species are also known from Croatia, Bosnia and Herzegovina and Montenegro (Radoman, 1975, 1983; Giusti & Pezzoli, 1980; Kabat & Hershler, 1993; Haase, 1994, 1996). WORMS (WoRMS, 2021) lists 93 records for the genus. Although many of them are now classified as belonging to other genera, still more than 40 nominal Belgrandiella species exist: Nearly all of them were described based only on the characters of the shell. Thus, relatively rich reports on the distribution of the nominal species—even of more than one living in sympatry at some caves or springs (e.g., Radoman, 1975, 1983; Bole, 1979; Sket, 1986, 1993, 1999; Bole et al., 1993; Žagar, 2012), are not a reliable source of information about real endemism, intra- and interspecies diversity, gene flow or speciation. Some molecular studies (Falniowski & Beran, 2015; Osikowski et al., 2018) have already reduced the number of valid species within this genus. The aim of the present paper is to check the inter- and intraspecies genetic diversity in (mostly Slovenian) Belgrandiella with mitochondrial cytochrome c oxidase subunit I—COI, nuclear histone H3, and RAPD analysis. The snails from caves, springs and interstitial aquifers are analysed to estimate the level of gene flow in order to determine whether these snails form a genetically uniform and widespread metapopulation (as Montenegrospeum), or they rather follow the highly endemic pattern found in Kerkia.
Material and methods
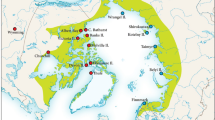
The snails were collected at 22 localities in Slovenia (Table 1, Figs. 1, 2). Seven of the localities (10–16) were in the cave Planinska jama (“jama” means cave in Slovenian). Planinska jama is the largest water cave in Slovenia, a karst cave that opens on the southern edge of Planinsko polje, near the village of Planina (Kogovšek et al., 2010). It consists of 6656 m of underground tunnels and is part of Postojna-Planinska jama System (hereafter referred to as PPCS). Into the Pivka channel of Planinska jama, flows the Pivka River that sinks in the cave Postojnska jama. Into the Rak channel of the Planinska jama, flows the Rak River that sinks into Tkalca jama. The Rak and Pivka Rivers both flow within their channels for about 2 km before they join together to form the Unica River about 500 m from the cave entrance. The side channels in both main channels are on upper level, they are older and mostly dry. They contain large amounts of cave sediments, especially alluvium brought in the cave by the underground Pivka River. The location of the debris indicates that parts of the cave have been completely filled at least once in the past. Most of the sediments are younger than 200,000 years, the cave developed over presumably more than two million years, with a more or less stable hydrological system between the Pivka Basin and Planinsko Polje (Kogovšek et al., 2010). Planinska jama is one of the underground habitats with high biodiversity, including the olm Proteus anguinus Laurenti, 1768, endemic species of amphipod Niphargus Schioedte, 1849, isopods Monolistra Gerstaecker, 1856, Asellus aquaticus cavernicolus Racovitza, 1925 and Titanethes albus (C. Koch, 1841), cave shrimp Troglocaris planinensis Birstein, 1948, cave hydrozoan Velkovrhia enigmatica Matjašič & Sket, 1971, and eight (Culver & Sket, 2000) mostly stygophile gastropod species (including terrestrial Zospeum Bourguignat, 1856 and pulmonate aquatic Ancylus fluviatilis O. F. Müller, 1774), whose empty, two to three millimeter-sized shells form dune-like thanatocenoses at some places. According to Sket (1999) in the Planinska jama PPCS 19, and according to Zagmajster et al. (2021) nine (in this seven Truncatelloid) aquatic gastropod species were found, and the number of troglobiotic and stygobiotic animal species approaches 120, similarly high number can be found only in Vjetrenica Cave in Bosnia and Herzegovina (Culver & Pipan, 2009).

Localities of the studied populations of Belgrandiella. Numbers correspond to localities list given in Table 1

Some of the studied localities: A locality 5, B locality 6, C locality 7, D locality 17, E locality 19, F locality 22
Also the material from earlier studies, collected at 14 other localities in Slovenia, Croatia, and Bosnia and Herzegovina (Table 1, Fig. 1) was considered. Empty shells and live specimens were obtained from caves, springs and wells by different sampling techniques: manual collection, washing of vegetation and substratum, the use of either hand-net or metal sieve, sometimes fitted with an extended handle (3 m) to enable sweeping through the water and substrate at the bottom. The Bou–Rouch method (Bou & Rouch, 1967) was used to sample an interstitial fauna inhabiting the spring or river hyporheic zone, at the cca. 50 cm depth. The sampling procedure was repeated 5 times at each site, and 20 L of a mixture of water, sediments and particulate organic matter were pumped each time. The mixtures were sifted through a hand-net of 500 μm mesh size and fixed in 80% analytically pure ethanol, replaced two times. The samples were sorted later, and the snails were put in fresh 80% analytically pure ethanol and kept at -20 C in a freezer.
The shells of the sequenced specimens were photographed with a CANON EOS 50D digital camera, under a NIKON SMZ18 microscope with dark field. Measurements of these shells, following the scheme given by Szarowska (2006; Fig. 44) were taken using ImageJ image analysis software (Rueden et al., 2017). The linear measurements were then logarithmically transformed; for angular measurements, the arcsine transformation was applied. Principal component analysis (PCA), based on the matrix of correlation, was computed, applying a descriptive, non-stochastic approach. The original observations were projected into PC space, to show the grouping of the specimens, without any classification given a priori. For PCA analysis ClustVis 2.0 web tool (Metsalu & Vilo, 2015) was used.
DNA was extracted from whole specimens; tissues were hydrated in TE buffer (3 × 10 min); then total genomic DNA was extracted with the SHERLOCK extraction kit (A&A Biotechnology), and the final product was dissolved in 20 μl of tris–EDTA (TE) buffer. The extracted DNA was stored at − 80 °C at the Department of Malacology, Institute of Zoology and Biomedical Research, Jagiellonian University in Kraków (Poland).
A fragment of mitochondrial cytochrome c oxidase subunit I (COI) and four nuclear fragments (18S ribosomal RNA – 18S, 28S ribosomal RNA – 28S, histone H3 – H3 and Internal Transcribed Spacer 2 – ITS2) were sequenced. Details of PCR conditions, primers used, and sequencing technique were given in Szarowska et al. (2016b), Hofman et al. (2022a, b). The Sanger sequencing was performed in Genomed Company in Warsaw, Poland. Sequences were initially aligned in the MUSCLE (Edgar, 2004) program accessed through MEGA 7 (Kumar et al., 2016). The correctness of the alignment was checked in BIOEDIT 7.2.5 (Hall, 1999), this program was also used to translate of sequences and check for reading frame and stop codons. Uncorrected p-distances were calculated in MEGA 7. The estimation of the proportion of invariant sites and the saturation test for the coding loci COI and H3 (Xia, 2000; Xia et al., 2003) were performed using DAMBE 7 (Xia, 2013). In the phylogenetic analysis additional sequences from GenBank were used (Table 2). The phylogenetic analysis was performed applying two approaches: Bayesian inference (BI) and maximum likelihood (ML). In the BI analysis, MRMODELTEST 2.3 (Nylander, 2004) was applied for each partition to find the best-fit model of nucleotide substitution.
The Bayesian analyses were run using MrBayes v. 3.2.3 (Ronquist et al., 2012) with defaults of most priors. Two simultaneous analyses were performed, each with 10,000,000 generations, with one cold chain and three heated chains, starting from random trees and sampling the trees every 1000 generations. The first 25% of the trees were discarded as burn-in. The analyses were summarised as a 50% majority-rule consensus tree. Convergence was checked in Tracer v. 1.5 (Rambaut & Drummond, 2009). FigTree v.1.4.4 (Rambaut, 2010) was used to visualise the trees. The maximum likelihood analysis was conducted in RAxML v. 8.2.12 (Stamatakis, 2014) using the ‘RAxML-HPC v.8 on XSEDE (8.2.12) tool via the CIPRES Science Gateway (Miller et al., 2010). Rapid bootstrap and autoMRE bootstopping criterion were used. We applied the GTR + G model, whose parameters were estimated for each partition by the RAxML (Stamatakis, 2014). It has been suggested that using the most parameter-rich model GTR + I + G instead of conducting thorough model selection leads to phylogenies with topologies (although not necessarily branch lengths) the same as those obtained when model selection is performed, or better, since the programs like ModelTest are sometimes definitely misleading (our observations). This implies that the standard practice of model selection before phylogeny inference is unnecessary when a complex and parameter-rich model is applied to nucleotide data (Abadi et al., 2019; our results), as long as the parameters are estimated separately for each partition. However, combining a gamma distribution (G) with a proportion of invariant sites (I) has been criticized (e.g., Sullivan et al., 1999; Mayrose et al., 2005; Yang, 2006; Jia et al., 2014; Stamatakis, 2014) mainly since some of the parameters of the + G and + I models cannot be optimized independently of each other, thus rather GTR + G model should be applied. Two methods of species delimitation, analysing COI sequences, were performed: Poisson Tree Processes (bPTP) (Zhang et al., 2013) and Automatic Barcode Gap Discovery (ABGD). The bPTP approach (bPTP) was run using the web server https://species.h-its.org/ptp/, with 100,000 MCMC generations, 100 thinning and 0.1 burn-in. We used RAxML output phylogenetic tree. The ABGD approach using the web server (https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html) and the default parameters. To infer haplotype network of the COI marker, we used a median-joining calculation implemented in NETWORK4 (Bandelt & Röhl, 1999). The parameters describing a genetic variation and other statistics were calculated with DnaSP 6.12.03 software (Librado & Rozas, 2009) and Arlequin v. 3.5 (Excoffier & Lischer, 2010). The latter program was used to analyze molecular variance (AMOVA). The geographical distances between the localities were calculated with Geographic Distance Matrix Generator v. 1.2.3 (Ersts, 2020). Mantel tests for association between the matrices and UPGMA clustering were performed with NTSYSpc 2.02 g (Rohlf, 1998). To apply the molecular clock, the data from COI were used. Two hydrobiids, Peringia ulvae (Pennant, 1777) and Salenthydrobia ferreri Wilke, 2003 (AF118302, Wilke & Davis 2000; AF449213, Wilke 2003), were used as outgroups. The divergence time of the lineage of Peringia (Atlantic) with the linage comprising Salenthydrobya + Achaiohydrobia (Mediterranean) was used to calibrate the molecular clock, with correction according to Falniowski et al. (2008). The likelihood ratio test (LRT) (Nei & Kumar 2000) were calculated with PAUP. The relative rate test (RRT) (Tajima 1993) was performed in MEGA. As Tajima’s RRTs and the LRT test rejected the equality of evolutionary rates throughout the tree for Belgrandiella, time estimates were calculated using a non-parametric rate smoothing (NPRS) analysis with the recommended Powell algorithm, in r8s v.1.7 for Linux (Sanderson 1997, 2003).
Details of randomly amplified polymorphic DNA (RAPD) analysis, primers used, and amplification technique were given in Falniowski et al. (2021a). The amplification products were separated in 2% agarose gel with ethidium bromide in TBE buffer. The banding patterns were visualised under UV light. The bands were scored visually, and those of similar molecular size (for the same primer) were assumed to be homologous. Each band was treated as an independent locus with two alleles: presence or absence of the band. The PopGen 32 software package (Yeh & Yang, 2000) was used to estimate the genetic variation. The following parameters were calculated: mean number of alleles per locus, effective number of alleles per locus, percentage of polymorphic loci and Nei’s (1973) gene diversity, the Shannon index. This same software was used to calculate the Nei’s genetic distances between populations.
Results
The shells of the studied Belgrandiella (Fig. 3) were either broadly conic or barrel-shaped, and highly variable in size. Some specimens were pigment- and eyeless (Figs. 3A, D, G-L, N-Q, and S-T), while the other showed no troglomorphism—their mantle was black pigmented, sometimes intensively, the eyes present. The principal component analysis (PCA) on seven shell biometric parameters (Fig. 4) demonstrated that the shell biometry of the six mOTUs (inferred from the mitochondrial COI, A-F, see below) slightly reflects their distinctness, since their variability ranges broadly overlap. The biggest ellipsoid contained specimens of B. kusceri (mOTU A), found in 23 from 36 (64%) populations. The PC1, reflecting mostly the size and describing about 65% of the total variance, ranged above three times more than each of the other five species. The PC2 and PC3, reflecting mostly differences in shell shape, showed different pattern. In the PC2 the range of the mOTU F was only slightly less wide than that of the mOTU A, and in PC3 the ranges were the same. For both PC1 and PC2 the highest character loadings were for the angles α (apex angle measured between the lines tangential to the spire) and β (angle between the body whorl suture and the line perpendicular to the columella). The loading of all the other characters on the PC3 were similarly low. For the PC2 higher values were computed for the body whorl breadth and aperture height, and stronger but negatively correlated—for the spire height. The three transects found for the RAPD in B. kusceri (Fig. 1) differ only in PC1 (Fig. 4). The transects III and IV did not overlap, but the transect II overlapped with both III and IV.

Shells of the sequenced Belgrandiella: A-R – mOTU A, S-T – mOTU E, U-Y – mOTU F, Z-AD – mOTU B; A – 1T1, B – 2O36, C – 1U40, D – 1P28, E – 2O34, F – 2O40, G – 1S54, H – 1S46, I – 2O2, J – 203, K – 2O5, L – 2D72, M – 1U58, N – 1S17, O – 1T2, P – 1S51, Q – 1P12, R – 1U55, S – 1S18, T – 1S43, U – 1W22, V – 1W23, W – 1W24, X – 1W12, Y – 1W13, Z – 1U84, AA – 1S35, AB – 2O15, AC – 1U80, AD – 2O72. The extraction numbers correspond to Figs. 5–7. Bar equals: 1 mm

Principal component analysis (PCA) on the shell of Belgrandiella, for different mOTUs (above) as well as for different groups within B. kusceri, distinguished with RAPD analysis for transects II, III, IV and locality 17 (below). Measured variables: a – shell height, b – body whorl breadth, c – aperture height, d – spire height, e – aperture breadth, α – apex angle measured between the lines tangential to the spire, β – angle between the body whorl suture and the line perpendicular to the columella
We obtained 56 new sequences of COI (457 bp, GenBank accession numbers OP804353-OP804408), 28 of H3 (310 bp, GenBank accession numbers OP824660-OP824687), 28 of the ITS2 (130 bp, GenBank accession numbers OP829059-OP829086), 28 of 28S (497 bp, GenBank accession numbers OP829115-OP829142), and 28 of 18S (401 bp, GenBank accession numbers OP829087-OP829114). The tests by Xia et al. (2003) revealed no saturation for COI and H3. Results from the substitution saturation analysis showed an ISS (0.65 for COI; 0.47 for H3) significantly smaller than the critical ISS value (ISSC: 0.94 for COI; 0.62 for H3). In all analyses, the topologies of the resulting phylograms were identical in both the ML and BI, for both COI and H3 sequences. For 18S, ITS2 and 28S all sequences were identical.
For all the sequences of cytochrome c oxidase subunit I (COI) for Belgrandiella (new and reference ones) we obtained 21 haplotypes with haplotype diversity (Hd) 0.895 ± 0.024. In total 61 sites were polymorphic (segregating) site and nucleotide diversity per site (Pi) was 0.0334 ± 0.0045. For H3 we obtained seven haplotypes with Hd 0.701 ± 0.051. In total 16 sites were polymorphic (segregating) site and Pi was 0.0117 ± 0.0013.
All the sequences of Belgrandiella belonged to one monophyletic clade in the tree based on histone H3 (Fig. 5). However, in the tree computed for COI (Figs. 6, 7), not all sequences of Belgrandiella formed a monophyletic clade, sister to Graziana alpestris (Frauenfeld, 1863), p-distance 0.11. There were four deviating sequences, identified as Belgrandiella cf. fontinalis (Schmidt, 1847) (mOTU C) and B. cf. koprivnensis Radoman, 1975 (mOTU D), which did not confirm their assignment to the genus Belgrandiella. In the clade of Belgrandiella inferred from COI, four mOTUs could be distinguished: B. kusceri (A. J. Wagner, 1914) (mOTU A, in 28 populations, although the COI sequences only for 23 of them) and B. cf. kuesteri (Boeters, 1970) (mOTU B). mOTU E was identified as B. crucis (Kuščer, 1928), and mOTU F as Belgrandiella. sp. Within the clade of Belgrandiella inferred from COI, the p-distances between mOTUs varied from 0.052 to 0.099 (Table 3). B. cf. koprivnensis and B. cf. fontinalis were more distinct from the other mOTUs (p-distance 0.082–0.121, Table 3). The species distinctness was confirmed by both used methods of species delimitation (for bPTP posterior probabilities were higher than 0.95). The geographic distribution of the mOTUs is shown in Fig. 8. While the mOTU A is distributed throughout the studied area, the mOTU B is restricted to the central Slovenia. The mOTU C is found at two localities in northern Slovenia and the mOTU D at a single, southernmost locality. Both the mOTUs E and F are also restricted to two Slovenian localities; the former to locations less than 20 m apart and the latter to localities 80 km apart. The estimated divergence times between the three main lineages are shown in Fig. 7.

Maximum likelihood H3 tree showing phylogenetic relationships between the studied snails. Bootstrap support (> 70%) and Bayesian posterior probabilities (0.95) are shown

Maximum likelihood COI tree showing phylogenetic relationships between the studied snails. Bootstrap support (> 65%) and Bayesian posterior probabilities (> 0.9) are shown. Results of the ABGD and bPTP analysis and division into the mOTUs are also shown

Maximum likelihood COI tree for haplotypes. Bootstrap support (> 70%) and Bayesian posterior probabilities (> 0.75) are shown. Results of the ABGD and bPTP analysis and division into the mOTUs are also shown. The estimated divergence times between the three main lineages are also indicated

Two of H3 haplotypes were characteristic for mOTU A, the other were characteristic each for one of the mOTUs. Individuals from five populations, not sequenced for the COI fragment, will cluster within mOTU A or C on the basis of H3 sequences (Table 1). For this locus all sequences belonged to one, well supported lineage. The p-distances between mOTUs varied from 0.012 to 0.034 (Table 3). From among 36 localities studied, at six (5, 6, 7, 23, 25 and 26) two species occurred in sympatry. It has to be stressed that interspecies distances for histone H3 were unusually high (Table 3). For COI AMOVA (Table 4) clearly assigned 94.34% of the total variance to the interspecies (between the mOTUs) differences. Within the mOTUs only 2.90% of the total variance was estimated, and nearly the same (2.76%) within populations.
The mOTU A, identified as B. kusceri, was most rich in the sequenced specimens (60) and in the studied populations (23), as well as most widely distributed geographically. Thus, it was possible to analyse its genetic structure, especially the pattern of the gene flow between the local (sub)populations. In this mOTU, 60 sequences represented 16 haplotypes, with Hd = 0.847 ± 0.036. Number of polymorphic (segregating) sites was 35 and nucleotide diversity (per site) 0.015 ± 0.003. Haplotypes network is shown in Fig. 9. Neighbouring haplotypes differed mainly by one substitution, two times by two substitutions. The geographic distribution of the haplotypes is shown in Fig. 10. The most numerous haplotype 1 was found at twelve localities across Slovenia. The most interesting population inhabited the cave Krška jama (locality 6), where five different haplotypes were found, four of them belonging to B. kusceri. Moreover, the latter haplotypes were unique for this population. In Žerovnica (locality 20), B. kusceri was represented by three haplotypes, and at the locality 8 it was represented by two haplotypes (different by five substitutions), 17, 12 and 19. Interestingly, in seven subpopulations (10–16) inhabiting the biggest water cave Planinska jama, only two haplotypes were found.

Haplotype network (non-hierarchical, no dichotomous tree) of the studied populations of Belgrandiella kusceri. The dots colors correspond to Fig. 10

Geographic distribution of the haplotypes of Belgrandiella kusceri (mOTU A)
Pairwise p-distances between the populations of B. kusceri in some cases approached 0.032. Mantel test, checking the association between the matrices of pairwise p-distance and geographic distance, thus testing the isolation-by-distance model, with 9999 permutations, resulted in p [random Z ≤ observed Z] = 0.6799, thus no significant association was found. The test between the matrices of geographic distance and pairwise FST (θ) (Table 5, Fig. 11), with 9999 permutations, resulted in p [random Z ≤ observed Z] = 0.0066, thus the association was highly significant. Pairwise theta (FST), Fisher’s estimator of the amount of structuring of a population into subpopulations, in general was high, with exception of populations sharing the same haplotype, then FST equalled 0, thus Nm infinity. In all the other cases Nm were low. Thus, this estimator derived from Fisher’s F-statistics, reflected low migration in subdivided population.

Pairwise FST vs geographic distance within Belgrandiella kusceri
In the RAPD analysis, each primer amplified from five to eight fragments, with an average of eight (overall 33 fragments), and among them 32 bands (96.97%) were polymorphic. Number of individuals and basic parameters of diversity are given in Table 6. Nei’s (1973) gene diversity ranged from 0.014 to 0.137, and the Shannon index ranged from 0.020 to 0.265. Mantel test of association between the matrices of geographic distance and Nei’s genetic distances (Table 7) with 9999 permutations, resulted in p [random Z ≤ observed Z] = 0.1531 thus not significant association was found. UPGMA clustering calculated for these Nei distances (Fig. 12) showed geographic pattern: (1) the cave Krška jama with two localities at the Krka River (populations 6–8, transect IV), (2) Žerovnica with Lipsenj (populations 18–22, transect III), (3) six populations in the Planinska jama (populations 10–16, transect II), and (4) one population from Rakov Škocjan (population 17). The geographical distribution of the RAPD bands for each of the primer are shown in Supp. Figure 1. It presents a similar differentiation, dependent on geographical distribution. In general, all three transects and population 17 differed in frequencies of particular bands and in populations there were often private bands. However, some bands were identified in all transects.

UPGMA dendrogram for populations of Belgrandiella kusceri using RAPD data. The Arabic letters indicates population numbers, Roman letters numbers of the transects, see Fig. 1
Discussion
Osikowski et al. (2018) partly revised the Balkan Belgrandiella, demonstrating its diversity overestimation, chaos in the species-level taxonomy and high variation in the shell morphology, with its ranges’ overlap. The same can be seen in our present materials. We follow the nomenclature proposed in that study. Belgrandiella cf. fontinalis (Schmidt, 1847) is mOTU C, as in Osikowski et al. (2018), and B.cf. koprivnensis Radoman, 1975 is mOTU D as in that study. B. kusceri (Wagner, 1914) is mOTU A as there, and B. cf. kuesteri (Boeters, 1970) is their clade B. The representatives of two other mOTUs, E and F, have not been studied by Osikowski et al. (2018). Considering descriptions, drawings and photographs, as well as the data on geographic distribution given by Kuščer (1928, 1932), Radoman (1975, 1983), Bole (1979), De Mattia (2005), Žagar (2012) and Sket & Stoch (2014), we identified mOTU E as B. crucis (Kuščer, 1928). In the case of the mOTU F, we have not been able to perform identification. Its shell has no distinct characteristics and may resemble either to B. fontinalis – but this name was previously assigned to the mOTU C (Osikowski et al., 2018), or to B. robusta Radoman, 1975. However, the histone H3 sequences of the topotypes of B. robusta, presented in Osikowski et al. (2018), were different from the ones of the mOTU F. Considering all the ambiguities in the species-level taxonomy of Belgrandiella and the nomenclatorial chaos, it seemed safer to leave this species unidentified.
The results of AMOVA strongly confirmed high interspecies differences parallelly to markedly low percent of variance explained by interpopulation and intrapopulation differences within the species. Somewhat unusually, the values of intra- and interpopulation variance proportion were nearly the same.
The genetic monomorphism in the stygobiotic organisms is often recorded (e.g., Culver & Pipan, 2009), but usually only a single or few specimens are available, allowing the possibility that the picture is biased at the expense of deficient material. However, sometimes the samples richer in number are available, and also then the monomorphism holds. Hofman et al. (2021) sequenced COI in as many as 13 specimens of Paladilhiopsis stellatus Hofman et al., 2021 from Zvezda Spring in Croatia, finding no polymorphism. Thus, the sympatric occurrence of as many as four unique haplotypes of B. kusceri in the cave Krška jama, in sympatry with B. crucis, is unusual and noteworthy. Three sympatric haplotypes at Žerovnica, and two at other four localities confirm rather common genetic polymorphism in Belgrandiella. Only two found haplotypes within the vast and microhabitat rich Planinska jama in contrast with five haplotypes from the small Krška jama, are hard to explain.
Low values of p-distance and low levels of intrapopulation polymorphism in B. kusceri may suggest rather high levels of gene flow, although selection cannot be excluded, and Nm estimates derived from FST were not high. As was stressed by Endler (1977), adaptive differences among populations can be maintained by natural selection even under high levels of gene flow, as well as selection may mimic effects of gene flow. This similarity may be also due to numerous stochastic factors, area effects, etc. No significant association between the p-distances and geographic distances may suggest rather the infinite-island model of interpopulation differentiation (Wright, 1978), expected for isolated populations, but the statistically significant association between pairwise FST and geographic distances suggests rather the stepping-stone model. If two populations did not share the same haplotype, the estimates of gene flow (Nm) derived from FST were low. If Nm > 1, the allele frequencies in the subpopulations remain homogenized (Wright, 1931, 1969). If Nm < 1 but still positive, an equilibrium based on the rate of mutation, migration, and genetic drift will be established. The haplotype network confirms low genetic diversification, but rather of dumbbell, not star-phylogeny pattern (Avise, 2000), with two starbursts, but close to each other – differing only by one mutation. Evidently there is a signature for a species that has expanded its range rather recently, but rather from more numerous founders than in the case of star-phylogeny. Longer time may also suggest the existence of two haplotypes in slowly evolving histone H3.
The RAPD results show finer resolution than COI, and the picture does not follow the one gained on the oxidase. Such dissimilarity was also observed in Planinska jama for Asellus aquaticus (Linnaeus, 1758) (Verovnik et al., 2003; Verovnik, 2012). Association between Nei and geographic pairwise distances was not significant, but the clustering presented nearly strictly geographic pattern.
In stygobites the pigmentation and eyes are sometimes still present (e.g., Culver & Pipan, 2009; Culver, 2012), but the pigmentation characterizing many populations of Belgrandiella may also suggest their stygophily. There are a few stygophile Balkan truncatelloid species like Sadleriana cavernosa Radoman, 1975 (Delicado, 2018), Litthabitella chilodia (Westerlund, 1886) or Radomaniola spp. (Falniowski et al., 2021b). However, Belgrandiella can be found in springs as single/a few specimens, rather overflooded from the subterranean waters, which rather contradicts possible stygophily of Belgrandiella.
Belgrandiella can be found in caves and springs, sometimes also as an element of the meiofauna, but in epikarst, only its empty shells can be found (Velkovrh, 1974). High intraspecies variability of the shell coupled with interspecies overlap of its variation forces caution with interpretation of all the faunistic records based solely on the shell morphology. Anyway, there are interesting observations on the geographic distribution of the aquatic subterranean snails, showing the same pattern as for the crustaceans (Sket, 1986, 1993). Species determinations in the latter group, based on several characters of their hard parts must be more reliable than the ones of the gastropods, based mostly on their shells, often even empty ones. Contrary to expectations, the species ranges in both groups do not follow the recent hydrographical units, but the presumed Pliocene ones. Karstification began in the end of Pliocene and continued through the Pleistocene (Matvejev 1961; Sket 1999). This may suggest that speciation took place in epigean streams before karstification, later the taxa invaded subterranean habitats and parallelly gained troglomorphic character states (many of them losing eyes and pigment, elongating appendages, etc.), but after hydrographical changes they have not changed their distribution ranges along the newly established water connections (Sket, 1986, 1993). The Pliocene lasted 5.33–2.58 Mya (Cohen et al., 2013), thus, assuming 1.83% per Mya estimate for the Hydrobiinae given by Wilke (2003), the time of divergence between our mOTUs would be 3.6–5.4 Mya (3.4 ± 1.7 Mya for our estimates) for the monophyletic clade inferred for COI (mOTUs A, B, E and F), and 4.4–5.8 Mya (4.7 ± 1.9 Mya and 4.5 ± 1.8 Mya for our estimates) for the other two mOTUs. Considering the non-ultrametricity of our molecular tree, as well as the rate of mutation not necessarily the same as in Hydrobia, these estimates are very rough, but still sufficient to confirm the hypothesis of Sket (1986, 1993) about the time and place of their speciation, preceding invasions of the subterranean habitats. Caves are often seen as analogues to the islands in the sea, long lasting in isolation. However, sometimes old species can be found on the islands much younger than these species (Szarowska et al., 2014). The case of Belgrandiella seems similar: already distinct, “good” species colonized caves. The pairwise p-distances within B. kusceri in several cases also clearly suggest the Pliocene as the time of divergence.
The competing “climate-relict” and “adaptive-shift” hypotheses have been proposed to explain the origins of cave organisms (Juan et al., 2010). According to the “climate-relict” model, preadapted ancestors took refuge (or simply colonized) in caves before the surface climate was altered by glaciation or aridification, and gradually adapted to the cave environment. Climatic oscillations caused local extinctions of the surface populations, leaving each relict population to evolve in allopatry, in a separate cave system. The “adaptive-shift” model supposes that preadapted ancestral species actively colonized caves to exploit novel resources and diverged under a gene flow scenario. Divergent selection between surface and subterranean habitats gradually overcame the homogenizing process of gene flow and eventually led to parapatric speciation (Zhang & Li, 2013). Molecular phylogenies are helpful in testing those hypotheses, since they allow reconstruction of sister relationships between cave and surface taxa (Rivera et al, 2002; Leys et al., 2003). The presence of either surface and cave populations in allopatry, or only relict subterranean species, or parapatric distributions of both surface and cave populations, may be criteria of distinction between the two hypotheses. In Belgrandiella, despite often pigmented populations, stygophily remains doubtful – the specimens in springs are few and they seem rather flooded from the subterranean waters. Nevertheless, Belgrandiella certainly does not reach the high abundance of e.g., Bythinella or Radomaniola that both inhabit also caves (Giusti & Pezzoli, 1977, 1980; Bichain et al., 2007; Falniowski et al., 2021b), thus should be classified as stygophiles. It has to stressed, however, that high densities of the latter are typical of springs, whereas Belgrandiella could be find in spring as single/a few specimens overflooded from the subterranean waters. Thus, a “climatic-relict “hypothesis seems more probable, but we should note again that the speciation preceded invasion of “isolated” caves (although earlier the snails may might inhabit interstitial habitats, subterranean but rather continuous) and later, in caves, only slight genetic changes took place.
Animals adapted to cave environments are traditionally thought to be highly geographically isolated because of their limited dispersal ability, resulting from limited physiological tolerances and, especially in the case of snails, physical limitations of their locomotion. The available data on the molecular genetics of the stygobiont gastropods did not confirm either the complete isolation of populations or their stability and longevity (Falniowski, 1992; Falniowski et al., 1998, 1999; Bilton et al., 2001; Brändle et al., 2005; Falniowski & Szarowska, 2011). For inhabitants of such miniature and changeable habitats (Szarowska, 2000) survival must depend on dynamic processes of colonization and recolonization events, coupled with short- or (in some cases) long-distance dispersal (Szarowska et al., 2016a, b). There are subterranean habitats that make migration between caves and springs possible – those are interstitial habitats, unconsolidated sediments bordering and underlying streams and rivers. They are neither rare nor discontinuous (Culver & Sket, 2000; Lamoreaux, 2004; Culver et al., 2009; Culver, 2012; Rysiewska et al., 2017; Haase et al., 2021). Falniowski et al. (2021a) reported high levels of gene flow, resulting in more than 200 km wide range of one haplotype of the truncatelloid Montenegrospeum bogici (Pešić et Glöer, 2012). On the other hand, in Kerkia inhabiting nearly the same territory as our Belgrandiella (and, in part, Montenegrospeum) the pattern was completely different (Hofman et al., 2022a, b). Like in Paladilhiopsis (Hofman et al., 2021), confirming the most widespread opinions on the stygobiont fauna, the intrapopulation genetic diversity does not (or nearly does not) exist, and the same concerns the intraspecies variation between populations. At the same time the differences between the species are high, the species have narrow geographic ranges restricted to the small geographic areas, and grouped also geographically, in three clades, thus the high level of endemism could be confirmed in Kerkia.
The pattern observed in our Belgrandiella species seems rather similar to the one described for Montenegrospeum, not for Kerkia, although in Belgrandiella kusceri (mOTU A) the geographic range is narrower – covering only about 42 km, and the levels of gene flow are lower. But as in Montenegrospeum, there are also some analogies in Belgrandiella distribution with the famous European Cave Salamander Proteus anguinus Laurenti, 1768 with wide and discontinuous range and cryptic species (Sket, 1997; Gorički & Trontelj, 2006), or the moitessieriid snail Bythiospeum (Richling et al., 2016).
Data Availability
Sequence data are also downloaded at GenBank; accession numbers OP829115-OP829142, OP829059-OP829086, OP829087-OP829114, OP804353-OP804408, OP824660-OP824687.
Code availability
Software application—BioEdit 7.2.5
References
Abadi, S., D. Azouri, T. Pupko & I. Mayrose, 2019. Model selection may not be a mandatory step for phylogeny reconstruction. Nature Communications 10: 934. https://doi.org/10.1038/s41467-019-08822-w.
Avise, J. C., 2000. Phylogeography, Harvard University Press, Cambridge, MA, The History and Formation of Species:
Bănărescu, P. M., 2004. Distribution Pattern of the Aquatic Fauna of the Balkan Peninsula. In Griffiths, H. I., B. R. Kryštufek & M. Jane (eds), Balkan Biodiversity, Pattern and Process in the European Hotspot, 203–217. https://doi.org/10.1007/978-1-4020-2854-0-12
Bandelt, H. J., P. Forster & A. Röhl, 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48.
Bernasconi, R. & A. Riedel, 1994. Mollusca. In Juberthie, C. & V. Decu (eds), Encyclopedia Biospeleologica I. Société Biospéléologie, Moulis: 54–61.
Bichain, J. M., P. Gaubert, S. Samadi & M. C. Boisselier-Dubayle, 2007. A gleam in the dark: Phylogenetic species delimitation in the confusing spring-snail genus Bythinella MoquinTandon, 1856 (Gastropoda: Rissooidea: Amnicolidae). Molecular Phylogenetics and Evolution 45: 927–941.
Bilton, D. T., J. R. Freeland & B. Okamura, 2001. Dispersal in freshwater invertebrates. Annual Review of Ecology and Systematics 32: 159–181.
Bole, J., 1979. Mehkužci Cerkniškega jezera i okolice. Acta Carsologica 8(3): 201–236.
Bole, J., B. Drovenik, N. Mršic & B. Sket, 1993. Endemic animals in hypogean habitats in Slovenia. Naše Jame [our Caves] 35(1): 43–55.
Bou, C. & R. Rouch, 1967. Un nouveau champ de recherches sur la faune aquatique souterraine. Comptes Rendus De L’académie Des Sciences, Paris, Série D 265: 369–370.
Brändle, M., I. Westermann & R. Brandl, 2005. Gene flow between populations of two invertebrates in springs. Freshwater Biology 50: 1–9.
Cohen, K. M., S. C. Finney, P. L. Gibbard & J.-X. Fan, 2013. The ICS International Chronostratigraphic Chart. Episodes 36: 199–204.
Culver, D. C., 2012. Mollusks. In White, W. B, & Culver D. C., (eds), Encyclopaedia of Caves (2nd Ed.). Academic Press, New York. Pp: 512–517.
Culver, D. C. & B. Sket, 2000. Hotspots of subterranean biodiversity in caves and wells. Journal of Cave and Karst Studies 62: 11–17.
Culver, D. C. & T. Pipan, 2009. The Biology of Caves and Other Subterranean Habitats, Oxford University Press, Oxford:, 254.
Culver, D. C. & T. Pipan, 2014. Shallow Subterranean Habitats. Ecology, Evolution and Conservation, Oxford University Press, Oxford:, 258.
Culver, D. C., T. Pipan & K. Schneider, 2009. Vicariance, dispersal and scale in the aquatic subterranean fauna of karst regions. Freshwater Biology 54: 918–929. https://doi.org/10.1111/j.1365-2427.2007.01856.x.
De Mattia, W., 2005. Slovenian hypogean molluscs (Prosobranchia: Hydrobiidae; Pulmonata: Ellobiidae) from Ljudevit Kuščer’s collection kept in the Natural History Museum of Trieste (Italy)Malakologische Abhandlungen. Staatliches Museum Für Tierkunde, Dresden 23: 55–66.
Delicado, D., 2018. A rare case of stygophily in the Hydrobiidae (Gastropoda: Sadleriana). Journal of Molluscan Studies 84: 480–485. https://doi.org/10.1093/mollus/eyy032.
Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797.
Endler, J., 1977. Geographic Variation, Speciation, and Clines, Princeton University Press, Princeton, NJ:
Ersts, P. J., 2020. Geographic Distance Matrix Generator (version 1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. Retrieved from: http://biodiversityinformatics.amnh.org/open_source/gdmg.
Excoffier, L. & H. E. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567.
Falniowski, A., 1992. The genus Bythinella Moquin-Tandon, 1855, in Poland (Gastropoda, Prosobranchia, Hydrobiidae). In: Gittenberger, E. & J. Goud (eds), Proceedings of the 9th International Malacological Congress. Unitas Malacologica, Leiden, 135–138.
Falniowski, A., 2018. Species Distinction and Speciation in Hydrobioid Gastropods (Mollusca: Caenogastropoda: Truncatelloidea). HSOA Archives of Zoological Studies 1(003): 1–6. http://dx.doi.org/https://doi.org/10.24966/AZS-7779/100003
Falniowski, A. & M. Szarowska, 2011. A new genus and new species of valvatiform hydrobiid (Rissooidea; Caenogastropoda) from Greece. Molluscan Research 31(3): 189–199.
Falniowski, A. & M. Szarowska, 2012. Sequence-based species delimitation in the Balkan Bythinella Moquin-Tandon, 1856 (Gastropoda: Rissooidea) with general mixed Yule coalescent model. Folia Malacologica 20: 111–120. https://doi.org/10.2478/v10125-012-0017-z.
Falniowski, A. & L. Beran, 2015. Belgrandiella A. J. Wagner, 1928 (Caenogastropoda: Truncatelloidea: Hydrobiidae): how many endemics? Folia Malacologica 23: 187–191. https://doi.org/10.12657/folmal.023.015
Falniowski, A., M. Szarowska, W. Fiałkowski & K. Mazan, 1998. Unusual geographic pattern of interpopulation variation in a spring snail Bythinella (Gastropoda, Prosobranchia). Journal of Natural History 32: 605–616.
Falniowski, A., K. Mazan & M. Szarowska, 1999. Homozygote excess and gene flow in the spring snail Bythinella (Gastropoda: Prosobranchia). Journal of Zoological Systematics and Evolutionary Research 37: 165–175. https://doi.org/10.1111/j.1439-0469.1999.tb00980.x.
Falniowski, A., M. Szarowska, I. Sirbu, A. Hillebrand & M. Baciu, 2008. Heleobia dobrogica (Grossu & Negrea, 1989) (Gastropoda: Rissooidea: Cochliopidae) and the estimated time of its isolation in a continental analogue of hydrothermal vents. Molluscan Research 28: 165–170.
Falniowski, A., M. Szarowska, P. Glöer & V. Pešić, 2012. Molecules vs morphology in the taxonomy of the Radomaniola/Grossuana group of Balkan Rissooidea (Mollusca: Caenogastropoda). Journal of Conchology 41: 19–36.
Falniowski, A., D. Georgiev, A. Osikowski & S. Hofman, 2016. Radiation of Grossuana Radoman, 1973 (Caenogastropoda: Truncatelloidea) in the Balkans. Journal of Molluscan Studies 82: 305–313. https://doi.org/10.1093/mollus/eyv062.
Falniowski, A., J. Heller, R. A. D. Cameron, B. Pokryszko, A. Osikowski, A. Rysiewska & S. Hofman, 2020. Melanopsidae (Caenogastropoda: Cerithioidea) from the eastern Mediterranean: another case of morphostatic speciation. Zoological Journal of the Linnean Society 20: 1–25. https://doi.org/10.1093/zoolinnean/zlz160.
Falniowski, A., B. Lewarne, A. Rysiewska, A. Osikowski & S. Hofman, 2021b. Crenobiont, stygophile and stygobiont molluscs in the hydrographic area of the Trebišnjica River Basin. ZooKeys 1047: 61–89.
Falniowski, A. & M. Szarowska, 2011a. Radiation and phylogeography in a spring snail Bythinella (Mollusca: Gastropoda: Rissooidea) in continental Greece. Annales Zoologici Fennici 48: 67–90. https://www.jstor.org/stable/23737066
Falniowski, A. & M. Szarowska, 2012. Phylogenetic position of Boleana umbilicata (Kuščer, 1932) (Caenogastropoda: Rissooidea). Folia Malacologica 20: 265–270.Falniowski, A. & M. Szarowska, 2013. Phylogenetic relationships of Dalmatinella fluviatilis Radoman, 1973 (Caenogastropoda: Rissooidea). Folia Malacologica 21: 1–7. https://doi.org/10.12657/folmal.021.001
Falniowski, A. & M. Szarowska, 2015. Species distinctness of Hauffenia michleri (Kuščer, 1932) (Caenogastropoda: Truncatelloidea: Hydrobiidae). Folia Malacologica 23: 193–195. https://doi.org/10.12657/folmal.023.016
Falniowski, A., V. Pešić, B. Lewarne, J. Grego, A. Rysiewska, A. Osikowski & S. Hofman, 2021a. Isolation and endemism in subterranean aquatic snails: unexpected case of Montenegrospeum bogici (Pešić et Glöer, 2012) (Gastropoda: Truncatelloidea: Hydrobiidae). Hydrobiologia 848: 4967–4990. https://doi.org/10.1007/s10750-021-04688-6.
Falniowski, A., V. Pešić & P. Glöer, 2014. Montenegrospeum Pešić et Glöer, 2013: a representative of Moitessieriidae? Folia Malacologica 22: 263–268. https://doi.org/10.12657/folmal.022.023
Giusti, F. & E. Pezzoli, 1977. Primo contributo alla revisione del genere Bythinella in Italia. Natura Bresciana, Annales Musei Civico Di Storia Naturale Di Brescia 14: 3–80.
Giusti, F. & E. Pezzoli, 1980. Gasteropodi, 2 (Gastropoda: Prosobranchia: Hydrobioidea, Pyrguloidea). Consiglio Nazionale delle Ricerche AQ/1/47. Guide per Il Riconoscimento Delle Specie Animali Delle Acque Interne Italiane 8: 1–67.
Glöer, P., 2002. Die Süswassergastropoden Nord- und Mitteleuropas. Bestimmungsschlussel, Lebensweise, Verbreitung. Die Tierwelt Deutschlands. Teil 73, Conchbooks, Hackenheim:, 327.
Gorički, Š & P. Trontelj, 2006. Structure and evolution of the mitochondrial control region and flanking sequences in the European cave salamander Proteus anguinus. Gene 378: 31–41.
Haase, M., 1994. Differentiation of selected species of Belgrandiella and the redefined genus Graziana (Gastropoda: Hydrobiidae). Zoological Journal of the Linnean Society 111: 219–246.
Haase, M., 1996. The radiation of spring snails of the genus Belgrandiella in Austria (Mollusca: Caenogastropoda: Hydrobiidae). Hydrobiologia 319: 119–129. https://doi.org/10.1007/BF00016880.
Haase, M., J. Grego, Z. P. Erőss, R. Farkas & Z. Fehér, 2021. On the origin and diversification of the stygobiotic freshwater snail genus Hauffenia (Caenogastropoda: Hydrobiidae) with special focus on the northern species and the description of two new species. European Journal of Taxonomy 775: 143–184. https://doi.org/10.5852/ejt.2021.775.1555.
Hall, T. A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98.
Hershler, R. & J. R. Holsinger, 1990. Zoogeography of North American hydrobiid snails. Stygologia 5: 5–16.
Hofman, S., J. Grego, A. Rysiewska, A. Osikowski & A. Falniowski, 2021. Two new species of the Balkan genus Paladilhiopsis Pavlović, 1913 (Caenogastropoda, Moitessieriidae). ZooKeys 1046: 157–176. https://doi.org/10.3897/zookeys.1046.64489.
Hofman, S., R. A. D. Cameron, M. Proćków, I. Sîrbu, A. Osikowski & A. Jaszczyńska A. M. Sokół & A. Falniowski, 2022a. Two new pseudocryptic species in the medium-sized common European land snails, Fruticicola Held, 1838; as a result of phylogeographic analysis of Fruticicola fruticum (O. F. Müller, 1774) (Gastropoda: Helicoidea: Camaenidae). Molecular Phylogenetics and Evolution 168: 107402.
Hofman, S., J. Grego, L. Beran, A. Jaszczyńska, A. Osikowski & A. Falniowski, 2022b. Kerkia Radoman, 1978 (Caenogastropoda: Hydrobiidae): endemism, apparently morphostatic evolution and cryptic speciation Molluscan Research. Molluscan Research. https://doi.org/10.1080/13235818.2022.2129943.
Illies, J. (ed), 1978. Limnofauna europaea, pp I-XVII, 1–532. Fischer G, Stuttgart.
Jia, F., N. Lo & S. Y. W. Ho, 2014. The impact of modelling rate heterogeneity among sites on phylogenetic estimates of intraspecific evolutionary rates and timescales. PLoS One 9: e95722. https://doi.org/10.1371/journal.pone.0095722.
Juan, C., M. T. Guzik, D. Jaume & S. J. B. Cooper, 2010. Evolution in caves: Darwin’s ‘wrecks of ancient life’ in the molecular era. Molecular Ecology 19: 3865–3880.
Kabat, A. & R. Hershler, 1993. The prosobranch snail family Hydrobiidae (Gastropoda: Rissooidea): review of classification and supraspecific taxa. Smithsonian Contributions to Zoology 547: 1–94.
Kogovšek, J., M. Petrič, N. Zupan-Hajna & T. Pipan, 2010. Planinska jama. DEDI - digitalna enciklopedija naravne in kulturne dediščine na Slovenskem, http://www.dedi.si/dediscina/89-planinska-jama.
Kumar, S., G. Stecher & K. Tamura, 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874.
Kuščer, L., 1928. Drei neue Höhlenschnecken. Glasnik Muzea Društva Sloveije, Ljubljana. 7: 50–51.
Kuščer, L., 1932. Höhlen und Quellenschneken aus dem Flussgebiet der Ljubljanica. Archiv Für Molluskenkunde 64: 48–62.
Lamoreaux, J., 2004. Stygobites are more wide-ranging than troglobites. Journal of Cave and Karst Studies 66: 18–19.
Leys, R., C. Watts, S. J. B. Cooper & W. F. Humphreys, 2003. Evolution of subterranean diving beetles (Coleoptera: Dytiscidae: Hydroporini, Bidessini) in the arid zone of Australia. Evolution 57: 2819–2834.
Librado, P. & J. Rozas, 2009. DnaSP v5 a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Matvejev, S., 1961. Biogeografija Jugoslavije (Biogeography of Yugoslavia), Monographies 9 Bioloski Institut Srbije NR, Beograd:
Mayrose, I., N. Friedman & T. Pupko, 2005. A gamma mixture model better accounts for among site rate heterogeneity. Bioinformatics 21: ii151–ii158. https://doi.org/10.1093/bioinformatics/bti1125.
Miller, M. A., W. Pfeiffer & T. Schwartz, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov., New Orleans, LA: 1–8.
Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America 70: 3321–3323.
Nei, M. & S. Kumar, 2000. Molecular evolution and phylogenetics, Oxford University Press, Oxford:
Nylander, J. A. A., 2004. MrModeltest v.2. Program distributed by the author, Evolutionary Biology Centre, Uppsala University, Uppsala:
Osikowski, A., S. Hofman, A. Rysiewska, B. Sket, S. Prevorčnik & A. Falniowski, 2018. A case of biodiversity overestimation in the Balkan Belgrandiella A. J. Wagner, 1927 (Caenogastropoda: Hydrobiidae): molecular divergence not paralleled by high morphological variation. Journal of Natural History 52: 323–344. https://doi.org/10.1080/00222933.2018.1424959.
Radoman, P., 1975. Specijacija u okviru roda Belgandiella i njemu srodnih rodova na Balkanskom poluostrvu. Glasnik Prirodnjačkih Muzeja, B Knjiga 30: 29–55.
Radoman, P., 1983. Hydrobioidea a superfamily of Prosobranchia (Gastropoda). I Systematics. Monographs of Serbian Academy of Sciences and Arts in Beograd 547, Department of Sciences 57: 1–256.
Rambaut, A., 2010. FigTree v1.3.1. <http://tree.bio.ed.ac.uk/software/figtree>.
Rambaut, A. & A. J. Drummond, 2009. Tracer v1.5. http://beast.bio.ed.ac.uk/Tracer.
Richling, I., Y. Malkowsky, Y. Kuhn, H.-J. Niederhöfer & H. D. Boeters, 2016. A vanishing hotspot – impact of molecular insights on the diversity of Central European Bythiospeum Bourguignat, 1882 (Mollusca: Gastropoda: Truncatelloidea). Organisms Diversity and Evolution 17: 67–85.
Rivera, M. A. J., F. G. Howarth, S. Taiti & G. K. Roderick, 2002. Evolution in Hawaiian cave isopods (Oniscidea: Philosciidae): vicariant speciation or adaptive shifts? Molecular Phylogenetics and Evolution 25: 1–9.
Rohlf, F. J., 1998. NTSYSpc, numerical taxonomy and multivariate analysis system. Version 2.0. Setauket, NY: Exeter Software.
Ronquist, F., M. Teslenko, M. P. Vander, D. L. Ayres, A. Darling, S. Höhna & J. P. Huelsenbeck, 2012. Mr. Bayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Rueden, C. T., J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena & K. W. Eliceiri, 2017. Image J2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18: 529. https://doi.org/10.1186/s12859-017-1934-z.
Rysiewska, A., S. Prevorčnik, A. Osikowski, S. Hofman, L. Beran & A. Falniowski, 2017. Phylogenetic relationships in Kerkia and introgression between Hauffenia and Kerkia (Caenogastropoda: Hydrobiidae). Journal of Zoological Systematics and Evolutionary Research 55: 106–117. https://doi.org/10.1111/jzs.12159.
Sanderson, M. J., 1997. A nonparametric approach to estimating divergence times in the absence of rate constancy. Molecular Biology and Evolution 14: 1218–1231. https://doi.org/10.1093/oxfordjournals.molbev.a025731.
Sanderson, M. J., 2003. R8s: inferring absolute rates of molecular evolution, divergence times in the absence of a molecular clock. Bioinformatics 19: 301–302. https://doi.org/10.1093/bioinformatics/19.2.301.
Sket, B., 1986. Evaluation of some taxonomically, zoogeographically, or ecologically interesting finds in the hypogean waters of Yugoslavia (in the last decades). 9. Congres International Espeleologie, Comunicate 2: 126–128.
Sket, B., 1993. Cave fauna and speleobiology in Slovenia. Naše Jame [our Caves] 35(1): 35–42.
Sket, B., 1997. Distribution of Proteus (Amphibia: Urodela: Proteidae) and its possible explanation. Journal of Biogeography 24: 263–280.
Sket, B., 1999. The nature of biodiversity in hypogean waters and how it is endangered. Biodiversity and Conservation 8: 1319–1338.
Sket, B. & F. Stoch, 2014. Recent Fauna of the Cave Križna jama in Slovenia. Mitteilungen Kommission Quartärforschung Österreichische Akademie Wissenschaften, 21: 45–55, Wien.
Sket, B., K. Paragamian & P. Trontelj, 2004a. A Census of the obligate subterranean fauna of the Balkan Peninsula. In Griffiths, H. I., B. Kryštufek & J. M. Reed (eds), Balkan Biodiversity: Pattern and Process in the European Hotspot Kluwer Academic Publishers, Dodrecht: 309–322.
Sket, B., P. Trontelj & C. Zagar, 2004b. Speleobiological characterization of the epikarst and its hydrological neighbourhood: its role in dispersion of biota, its ecology and vulnerability. In: Jones, W.K., Culver, D.C., and Herman, J.S. (eds.), Epikarst, Special Publication 9, Karst Waters Institute, Leesburg, Virginia: 104–113.
Stamatakis, A., 2014. RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. https://doi.org/10.1093/bioinformatics/btu033.
Sullivan, J., D. L. Swofford & G. J. P. Naylor, 1999. The effect of taxon sampling on estimating rate heterogeneity parameters of maximum-likelihood models. Molecular Biology and Evolution 16: 1347–1356. https://doi.org/10.1093/oxfordjournals.molbev.a026045.
Szarowska, M., 2000. Environmental threats and stability of Bythinella populations in South Poland (Gastropoda: Prosobranchia: Hydrobioidea). Malakologische Abhandlungen, Staatliches Museum Fr Tierkunde Dresden 20: 93–986.
Szarowska, M., 2006. Molecular phylogeny, systematics and morphological character evolution in the Balkan Rissooidea (Caenogastropoda). Folia Malacologica 14: 99–168. https://doi.org/10.12657/folmal.014.014.
Szarowska, M., S. Hofman, A. Osikowski & A. Falniowski, 2014. Divergence preceding Island formation among Aegean insular populations of the freshwater snail genus Pseudorientalia (Caenogastropoda: Truncatelloidea). Zoological Science 31: 680–686.
Szarowska, M., A. Osikowski, S. Hofman & A. Falniowski, 2016a. Does diversity patterns of the spring-inhabiting snail Bythinella (Gastropoda, Bythinellidae) on the Aegean Islands reflect geological history? Hydrobiologia 765: 225–243. https://doi.org/10.1007/s10750-015-2415-x.
Szarowska, M., A. Osikowski, S. Hofman & A. Falniowski, 2016b. Pseudamnicola Paulucci, 1878 (Caenogastropoda: Truncatelloidea) from the Aegean Islands: a long or short story? Organisms Diversity and Evolution 16: 121–139. https://doi.org/10.1007/s13127-015-0235-5.
Tajima, F., 1993. Simple methods for testing molecular clock hypothesis. Genetics 135: 599–607. https://doi.org/10.1093/genetics/135.2.599.
Velkovrh, F., 1974. Razsirjenost gastropodov po drobnih razpokah v krasu (The distribution of Gastropoda in small karstic fissures): Naše jame [Our caves] 15, 77–81.
Verovnik, R., 2012. Asellus aquaticus: a model system for historical biogeography. In Culver, D. C. & W. B. White (eds), Encyclopaedia of Caves 2nd ed. Academic Press, Amsterdam: 30–36.
Verovnik, R., B. Sket, S. Prevorčnik & P. Trontelj, 2003. Random amplified polymorphic DNA diversity among surface and subterranean populations of Asellus aquaticus (Crustacea: Isopoda). Genetica 119: 155–165.
Wagner, A., 1928. Studien zur Molluskenfauna der Balkanhalbinsel mit besonderer Berücksichtigung Bulgariens und Thraziens, nebst monographicher Bearbeitung einzelner Gruppen. Annales Zoologici Musei Polonici Historiae Naturalis Warszawa 6: 263–399.
Wilke, T., 2003. Salenthydrobia gen. nov. (Rissooidea: Hydrobiidae): a potential relict of the Messinian salinity crisis. Zoological Journal of the Linnean Society 137: 319–336. https://doi.org/10.1046/j.1096-3642.2003.00049.x.
Wilke, T. & G. M. Davis, 2000. Infraspecific mitochondrial sequence diversity in Hydrobia ulvae and Hydrobia ventrosa (Hydrobiidae: Rissoacea: Gastropoda): Do their different life histories affect biogeographic patterns and gene flow? Biological Journal of the Linnean Society 70: 89–105. https://doi.org/10.1111/j.1095-8312.2000.tb00202.x.
Wilke, T. & A. Falniowski, 2001. The genus Adriohydrobia (Hydrobiidae: Gastropoda): Polytypic species or polymorphic populations? Journal of Zoological Systematics and Evolutionary Research 39: 227–234. https://doi.org/10.1046/j.1439-0469.2001.00171.x.
Wilke, T., G. M. Davis, A. Falniowski, F. Giusti, M. Bodon & M. Szarowska, 2001. Molecular systematics of Hydrobiidae (Gastropoda: Rissooidea): testing monophyly and phylogenetic relationships. Proceedings of the Academy of Natural Sciences of Philadelphia 151: 1–21.
WoRMS, Editorial Board, 2021. World Register of Marine Species. Available from https://www.marinespecies.org at VLIZ. Accessed 2021–12–27. doi:https://doi.org/10.14284/170
Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159.
Wright, S., 1969. Evolution and the Genetics of Populations, Vol. 2. University of Chicago Press, Chicago, The Theory of Gene Frequencies:
Wright, S., 1978. Evolution and the genetics of populations, Vol. 4. University of Chicago Press, Chicago, Variability within and among natural populations:
Xia, X., 2000. Data analysis in molecular biology and evolution, Kluwer Academic Publishers, Boston, Dordrecht & London:
Xia, X., 2018. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Molecular Biology and Evolution 35: 1550–1552.
Xia, X., Z. Xie, M. Salemi, L. Chen & Y. Wang, 2003. An index of substitution saturation and its application. Molecular Phylogenetics and Evolution 26: 1–7.
Yang, Z., 2006. Computational Molecular Evolution. Oxford University Press, New York, 376 pp. https://doi.org/10.1093/acprof:oso/9780198567028.001.0001
Yeh, F. C. & R. Yang, 2000. POPGENE Version 1.32. Dept. of Renewable Resources. Univ. of Alberta. https://sites.ualberta.ca/~fyeh/popgene.html.
Žagar, S.,2012. Fauna polžev v izvirih okolice Cerkniškega jezera. Dipl. delo: Ljubljana, Univerza v Ljubljani, Biotehniška fakulteta, Oddel za biologijo.
Zagmajster, M., S. Polak & C. Fišer, 2021. Postojna-Planina Cave System in Slovenia, a Hotspot of Subterranean Biodiversity and a Cradle of Speleobiology. Diversity 13: 271. https://doi.org/10.3390/d13060271.
Zhang, Y. & S. Li, 2013. Ancient lineage, young troglobites: recent colonization of caves by Nesticella spiders. BMC Evolutionary Biology 13: 183.
Zhang, J., P. Kapli, P. Pavlidis & A. Stamatakis, 2013. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 29: 2869–2876.
Acknowledgements
The study was supported by a grant from the National Science Centre 2017/25/B/NZ8/01372 to Andrzej Falniowski.
Funding
The study was supported by a grant from the National Science Centre 2017/25/B/NZ8/01372.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Handling Editor: Manuel Lopes-Lima
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaszczyńska, A., Falniowski, A., Prevorčnik, S. et al. Isolation and endemism in the subterranean aquatic snails of the genus Belgrandiella A. J. Wagner, 1928 (Caenogastropoda: Truncatelloidea: Hydrobiidae). Hydrobiologia 850, 4089–4113 (2023). https://doi.org/10.1007/s10750-022-05106-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-022-05106-1