
Abstract
The blue and red shrimp Aristeus antennatus has been intensively exploited by trawling fishery in the Western Mediterranean Sea. Implementation of an adequate management plan needs complete genetic data of the non-spawning and spawning grounds of the species. During the reproductive period, the species forms spawning aggregations in the middle slope, mainly constituted of mature females carrying spermatophores. Seven spawning females’ grounds in the Western Mediterranean Sea from Gulf of Lions (France) to Cabo de Palos (Spain) were sampled during two consecutive years, 2016 and 2017. This study assessed for the first time the levels of genetic diversity within spawning grounds, the genetic divergence among them and estimated the degree of temporal genetic stability using multilocus genotyping. Our results showed a high connectivity of the spawning females’ grounds that remains stable during the two analysed years, explained by passive larval dispersal, together with juveniles and adults’ migration. We advise that a future management plan for A. antennatus fishery should consider the Mediterranean Subarea GSA6 as a single unit, but it should be combined with local monitoring in order to ensure the sustainable exploitation of the populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2002, the Council of the European Union emphasized the need of conducting a sustainable exploitation of fishery resources in order to ensure their long-term viability (European Council, 2002). The inclusion of genetic results in management advice is crucial to achieve that goal, especially when it is combined with life-traits and ecological information (Allendorf et al., 2008; Waples et al., 2008; Waples & Naish, 2009). Indeed, the exploitation of a fishery resource can generate, among others, a loss of genetic diversity within subpopulations, and a modification of fine-scale genetic structure and gene flow. Those genetic changes could lead to a reduction in the recovery of the population or increase extinction risks, produce a decrease in biomass and a reduction of the sizes at first maturity (Allendorf et al., 2008). Genetic markers assist in identifying the species' conservation units, in other words the populations, and to estimate their structure, connectivity and genetic diversity (Hauser & Carvalho, 2008). In addition, genetic studies allow to improve the assessment or development of management strategies to ensure the sustainability of exploited marine resources (Ward, 2000).
Furthermore, the population structure of a marine species could be temporally unstable due to several factors, such as processes that modify the ecosystem, sweepstake reproductive success or the individuals’ behaviour (Pilling et al., 2008; Waples & Naish, 2009). Hence, an accurate research about the identification of the genetic structure in marine species should include temporal replicates (Waples & Naish, 2009). In addition to molecular data, it is also important to collect other biological data, such as life-history traits (e.g. the size), in order to obtain integrative information on the species (Carvalho & Hauser, 1994). It has been pointed out that a fishery should not show a tendency in the annual variation in size composition of catches so that it could be considered sustainable, since a reduction of individuals’ size over time has been indicated as a possible consequence of overfishing (Leitão, 2019).
Aristeus antennatus (Risso, 1816) (Crustacea, Aristeidae) (En—Blue and red shrimp; Fr—Crevette rouge; Sp—Gamba roja) is distributed across the whole Mediterranean Sea, although it increases westwards (Sardà et al., 2004; Fernández et al., 2011). In response to the overexploitation of A. antennatus in the Western Mediterranean, the General Fisheries Commission for the Mediterranean Sea decided to include this species into the priority list for action (FAO, 2006), urging to a reduction of the fishing effort in the Balearic Islands and the Eastern coast of Spain (Mediterranean Subareas GSA5 and GSA6, respectively). In fact, even though A. antennatus in these areas has been considered one of the main marine resources where it has been intensively exploited by trawling fishery (Massutí et al., 2008), a management plan specific for the species which integrates genetic information with traditional methods has not been developed yet. Nowadays, the fishery of the species is carried out following general guidelines for trawling fisheries (European Council, 2019). To implement an adequate management plan for A. antennatus, it is necessary to identify the population structure with appropriate spatial and temporal replicates (Hauser & Carvalho, 2008; Waples & Naish, 2009), thus ensuring that genetic patterns are stable over time and not caused by sampling artefacts (Waples, 1998).
The blue and red shrimp is the most eurybathic species in the Mediterranean Sea, inhabiting the muddy bottoms from 80 m to nearly 3000 m of depth (Sardà et al., 2004). In the GSA6, the highest abundances have been found in Palamós, Blanes, Dénia and Santa Pola (Esteban, 2018). Adult females are more abundant at depths shallower than 1000 m, whereas the proportion of adult males and juveniles increase in deeper layers (below 1000 m) (Sardà et al., 2003, 2004). The spawning period takes place from late May to late September, with maximum values between June and August (Demestre, 1995). Spawning aggregations are formed in the middle slope (600–800 m depth) during that period, mainly composed of spawning females carrying one or more spermatophores (Sardà et al., 1997; Gorelli et al., 2014; Planella et al., 2019). Since fishery is performed between depths of 400 and 800 m (Carbonell et al., 1999; Maynou, 2008), catches are essentially constituted of females (Gorelli et al., 2014). A. antennatus presents sex dimorphism, where females show higher size than males, estimated by the cephalothorax length (CL) (Sardà et al., 2004; Deval & Kapiris, 2016). Sexual maturity in females is indicated according to the stage of gonad maturity, and 26 mm CL is the size at first maturity, which corresponds to age-1 individuals (Demestre & Martín, 1993; Carbonell et al., 2008).
A recent study using microsatellite markers in A. antennatus indicated a high level of genetic connectivity among fishing grounds along the Mediterranean Subarea GSA6 (from Gulf of Lions, France, to Cabo de Palos, Spain), supported mainly by the low level of population differentiation (i.e. low FST values) and the high migration rates (Agulló et al., 2020). In that study, seven fishing grounds (Port de la Selva, Roses, Palamós, Blanes, Vilanova i la Geltrú, Dénia and Santa Pola) were sampled during winter seasons of 2016 and 2017, when captures are mostly composed of small-sized specimens (16–34 mm CL, cohorts 1 + and 2 +) and the sex ratio is 1:1 (Carbonell et al., 1999; Agulló et al., 2020). A small but significant regional structure was suggested among 2017 samples, and also local temporal genetic fluctuations between 2016 and 2017, mainly due to the oceanographic singularity at the Blanes fishing ground.
Performing studies during spawning season in addition to non-breeding season is critical because the maximum integrity of the stocks appears during the reproductive period (Reiss et al., 2009). During the spawning period, stocks are likely to present higher genetic differences among them than during non-spawning period, even over short geographical distances (Hauser & Carvalho, 2008 and references therein). Several marine species have been analysed during the spawning period, such as the short-finned squid Illex argentinus (Castellanos, 1960) (Roldán et al., 2014), the white-streaked grouper Epinephelus ongus (Bloch, 1790) (Nanami et al., 2017) and the deep-water velvet shrimp Metapenaeopsis sibogae (De Man, 1907) (Rahman & Ohtomi, 2017). In A. antennatus, spawning aggregations are formed during summer, involving females from 1 + to older than 5 + cohorts (Carbonell, 2005) so it would be relevant to evaluate the population genetics during that season.
Here, we expand our former study focussing our attention on summer spawning females. The genetic diversity and divergence of the spawning grounds of the blue and red shrimp were assessed using multilocus genotyping. The studied area covered the Mediterranean Subarea GSA6 by sampling two consecutive years, 2016 and 2017. A multilocus study sampling spawning females would allow to test the population structure and the stability of the genetic pattern in the same years as Agulló et al. (2020). If the genetic pattern was not maintained, it could be due to several reasons, such as sex-biassed dispersal and mesoscale features. Finally, the integration of our outcomes into future management plans could improve the species’ fishery, making it more sustainable, especially in the medium and long term.
Materials and methods
Sampling
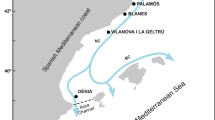
A total of 1400 adult females of A. antennatus were obtained from 7 Western Mediterranean locations (GSA6: Port de la Selva, Roses, Palamós, Blanes, Vilanova i la Geltrú, Dénia and Santa Pola), with a North–South distance of 719 km (Fig. 1) in two consecutive years (2016 and 2017). All samples were captured by commercial trawling vessels during July, and were provided by local fishermen on the date of capture (Table 1). For each individual, CL was measured from the orbital margin to the mid-posterior edge of the carapace, using a digital vernier calliper. Spermatophores were recorded on the thelycum of almost every captured female, which were also sexually mature according to the macroscopic external coloration of the ovary (Demestre & Fortuño, 1992). Analysis of variance (ANOVA) and Tukey post hoc tests were performed to compare the individuals’ CL among samples using the GraphPad Prism version 8.4.1 (GraphPad Software, San Diego, CA, USA). Besides, an unpaired two-sided Student’s t test was performed to compare all the individuals’ CL between the two temporal collections.

Sampling locations of Aristeus antennatus in the Western Mediterranean Sea. Further details are given in Table 1
DNA extraction, microsatellite amplification and genotyping
A portion of muscle tissue from each specimen was stored in 70% ethanol. Total genomic DNA extraction was performed according to the phenol: chloroform procedure outlined in Fernández et al. (2013). After isolation, DNA extractions were resuspended in ddH20 and stored at − 20 °C. Twelve previously reported microsatellite loci (Aa123, Aa1255, Aa138, Aa1444, Aa496b, Aa667, Aa681, Aa751, Aa956, Aa1061, Aa1195 and Aa818; Heras et al., 2016) were amplified with three PCR multiplex and one PCR singleplex, following the conditions published in Planella et al. (2019). PCR products were visualized in an automatic sequencer (ABI PRISM 3730xl, Applied Biosystems, Foster City, CA) at the sequencing unit of the University of Santiago de Compostela (Campus Lugo, Spain). All genotypes were obtained by allele scoring with GeneMapper 4.0 software (Applied Biosystems, Foster City, CA). GeneScan 500 LIZ Dye Size Standard (Applied Biosystems) was used as internal standard. Individuals genotyped for < 8 loci were excluded from further consideration, leaving a total of 1343 individuals (95.9% of sampled specimens) suitable for genetic statistical analyses.
Genetic data analysis
The software Genepop v. 4.6 (Rousset, 2008) was used to estimate allele frequencies, observed (Ho) and expected (He) heterozygosities, inbreeding coefficient (FIS) (Weir & Cockerham, 1984), to test for deviations from Hardy–Weinberg equilibrium (HWE), and for linkage disequilibrium between pairs of loci. The last two tests were performed with 10,000 dememorizations, 100 batches and 5000 iterations per batch, and P values were adjusted for multiple comparisons using the Bonferroni correction (Rice, 1989). Number of alleles per locus (NA) and allelic richness (AR) were computed using FSTAT v. 2.9.3 (Goudet, 2001). Micro-Checker v. 2.2.3 (Van Oosterhout et al., 2004) was performed to evaluate the frequency of possible null alleles with 1000 randomizations.
Pairwise FST values among all locations and hierarchical analysis of molecular variance (AMOVA; Excoffier et al., 1992) were performed using Arlequin v. 3.5 (Excoffier & Lischer, 2010). The significance of F-statistics was assessed by permutation tests with 10,000 replicates. FreeNA (Chapuis & Estoup, 2007) was carried out to correct pairwise FST values for null alleles. Hierarchical models of AMOVA were used to assess the partitioning of genetic diversity within and among spawning grounds in 2016 and 2017, as well as the temporal component (2016 vs 2017). Additionally, we calculated pairwise FST between sampling campaigns (winter 2016, summer 2016, winter 2017 and summer 2017) for each fishing ground to assess the seasonal component using winter female data from Agulló et al. (2020).
The effective population size (Ne) was calculated for the fourteen samples using the bias-corrected version of the method based on linkage disequilibrium (Hill, 1981; Waples, 2006) as implemented in NeEstimator v. 2.1 (Do et al., 2014), and 95% confidence intervals were determined using the non-parametric Jack-knife method. Given the minimum sample size of 77 (VIG16 in Table 2), a critical allele frequency (Pcrit) of 0.01 was applied to ensure that Pcrit > 1/2 N, where N represents the total sample size, in order to avoid single copy alleles in the analysis.
Pearson’s chi-square and Fisher exact tests were carried out in the software POWSIM v. 4.1 (Ryman & Palm, 2006) to assess the statistical power of our set of loci to detect significant genetic differentiation for the number of loci, the allele frequencies, and sample sizes (1000 dememorizations, 100 batches and 1000 iterations) and 1000 replicates. The power of the tests for detecting the defined level of genetic divergence was interpreted by the percentage of significant outcomes. Different values for FST were explored by changing the number of generations of drift (t).
Isolation by distance (IBD) among samples within temporal collection was evaluated testing the correlation between genetic (linearized as FST/(1−FST); Rousset, 1997) and geographical distances (measured as the shortest coastline between two locations in kilometres using Google Earth) with a Mantel test with 1000 permutations by NTSYSpc v. 2.1 (Rohlf, 1993).
We investigated the direction and magnitude of relative migration between samples within temporal collections with div-Migrate online (Sundqvist et al., 2016). For each temporal collection, a relative migration network was generated using the effective number of migrants (Nm) estimator. A filter threshold was set to 0.4 in both 2016 and 2017.
Results
Statistical analysis of the cephalothorax length
Despite all individuals were considered to belong to the medium-sized commercial category by the fishermen, the total CL’s range varied from 24.63 mm (in SPO16) to 47.18 mm (in PAL17) (Fig. 2). The Tukey’s post hoc test indicated significant differentiation in 71 out of 91 pairwise comparisons (Supplementary Table S1). Furthermore, the unpaired t test performed between the two temporal 2016 and 2017 collections showed a significant differentiation (t = 11.29, degree of freedom = 1398, P value < 0.0001).

Mean cephalothorax length (CL) per sample, with standard deviation. Each colour represents a location. Sample codes as in Table 1
Diversity within locations
The number of alleles per locus (NA) ranged between 2 and 23 in 2016 samples and from 2 to 24 in 2017 ones (Supplementary Table S2). The average NA was 8.8 in 2016 and 9.1 in 2017 (Table 2). The mean allelic richness (AR) was 8.2 in 2016 and 8.9 in 2017. The mean observed heterozygosity (Ho) was 0.461 in 2016, whereas in 2017 it was 0.479. The mean expected heterozygosity (He) was 0.628 in 2016 and 0.635 in 2017 (Table 2). Significant linkage disequilibrium was detected in the pair of loci Aa1444 and Aa681 in SPO17 after Bonferroni correction (P < 0.0001), but not in the other 13 samples.
Only three out of twelve loci (Aa138, Aa496b and Aa1195) showed no departure from the Hardy–Weinberg equilibrium (HWE) in any sample (Supplementary Table S2). The fourteen samples presented a significant deviation from HWE after the Bonferroni correction due to heterozygote deficit, as indicated by the observed positive FIS values (Table 2). This fact could be a consequence of null alleles. Estimated allele frequencies for null alleles were generally low and they were not consistent in all loci and across samples (Supplementary Table S2). Only the locus Aa818 was discarded from all subsequent analyses because it showed null alleles frequency higher than 0.2 in four samples. Additionally, significant departures from HWE might result from pooling specimens from distinct cohorts.
Geographical and temporal variation
Five out of the fourteen samples had an estimated infinite Ne, whereas in three it was close to 500 (Table 2), so Ne = 500 and Ne = 10,000 were selected to assess the statistical power of our loci to detect genetic differentiation. The evaluation of the statistical power estimated a high probability (> 99%) of detecting structure for 0.001–0.003 FST values, suggesting that our test would detect a real population structure if true estimates of FST were at this level. Nevertheless, AMOVA results did not detect significant genetic divergence either in the 2016 or in 2017 (FST = 0.0013, P > 0.05 and FST = 0.0003, P > 0.05, respectively) (Table 3). Likewise, analyses of pairwise genetic differentiation between each pair of samples within each temporal collection showed no significant FST values ranging from 0.0000 to 0.0028 in both collections (Table 4). In addition, there were no substantial differences in estimated FST values correcting, or not, for the presence of null alleles.
The AMOVA performed to compare the two temporal collections revealed no significant variation between them (FCT = 0.0002, P > 0.05) (Table 3). Furthermore, no pairwise FST differed significantly from zero when comparing the two temporal samples of each location (P > 0.05) (Supplementary Table S3). On the other hand, none of the FST between consecutives sampling campaigns (winter 2016, summer 2016, winter 2017 and summer 2017) at each of the seven fishing grounds was significant (Table 5) and no significant genetic differences were observed throughout the entire study period (winter 2016 vs. summer 2017).
No association between genetic differentiation and geographical distance was detected either in 2016 (r = − 0.1925, P = 0.228) or in 2017 samples (r = − 0.2861, P = 0.138). Results of migration indicated a high level of genetic connectivity among all samples at the two summer seasons, without a pattern relating the level of connectivity with geographical proximity (Fig. 3).

Relative migration networks estimated by div-Migrate-online. Circles represent samples, while arrows indicate the direction and magnitude of relative migration levels using Nm estimator. Thicker arrows indicate stronger migration. Sample codes as in Table 1. Networks are shown for the two temporal collections, a 2016 and b 2017
Discussion
Diversity within locations
The mean observed and expected heterozygosities are in accordance with the previous studies using the same set of microsatellite loci and the same species (Ho = 0.470, He = 0.632, Table 2) (Ho = 0.458, He = 0.628, Heras et al., 2019; Ho = 0.443, He = 0.611 recalculated excluding Aa421, Planella et al., 2019; Ho = 0.468, He = 0.633, Agulló et al., 2020). The mean number of alleles per locus (NA = 9.0, Table 2) was slightly higher than the values obtained in Heras et al. (2019) (NA = 7.8) and Planella et al. (2019) (NA = 7.9 recalculated excluding Aa421) due to the higher sample size of the present study (N = 494 in Heras et al., 2019; N = 111 in Planella et al., 2019). Indeed, the mean NA was almost the same when analysing similar sample size in winter season (N = 1386, NA = 9.1, Agulló et al., 2020).
The heterozygote deficit (i.e. FIS positive values), which is responsible for the deviations from HWE found across the fourteen samples, is a common feature on penaeids (Heras et al., 2016 and references therein). One possible explanation for the FIS positive values is the presence of null alleles (Supplementary Table S2). On the other hand, heterozygote deficits can reflect Wahlund effects resulting from the collection of differentiated subpopulations or cohorts in the same sample (Addison & Hart, 2005). Studied individuals were classified by the vessels’ crew on board according to a commercial category (medium-sized shrimp), instead of according to a modal size class related to a specific cohort. Furthermore, the mean size of individuals in the medium-sized category was slightly different among vessels’ crew. In fact, the mean CL was also significantly different among several samples (Supplementary Table S1), even between the two year collections at some locations (Fig. 2 and Supplementary Table S1). Females CL at modal sizes and cohorts were reviewed by Carbonell (2005), who suggested that females of around 30 mm CL would be classified as cohort 2 + , whereas females between 38 and 40 mm CL would be classified as 3 + . In this study, the mean CL among all samples was 37.04 ± 3.42, which confirms the pooling of different cohorts in our samples.
Genetic connectivity pattern and temporal stability
The main objective of this study was to assess for the first time the divergence of the spawning females’ grounds of A. antennatus during the reproductive period in the Mediterranean Subarea GSA6. Our results showed an overall regional genetic homogeneity confirming a high genetic connectivity among females between spawning aggregations (Fig. 3, Tables 3 and 4), as recently observed from samples captured during the winter season at the same fishing grounds, when smaller males and females contributed to the fishery (Agulló et al., 2020, but see below for local divergences). Our findings also agree with the previous studies indicating high genetic homogeneity among A. antennatus populations in the Western and Central Mediterranean Sea applying different genetic markers such as allozymes (Sardà et al., 1998), mitochondrial DNA (Maggio et al., 2009; Roldán et al., 2009; Sardà et al., 2010; Fernández et al., 2011; Marra et al., 2015) and microsatellites (Cannas et al., 2012; Heras et al., 2019).
The high level of genetic connectivity that it has been reported in the species is probably due to passive dispersal by oceanographic currents and active migration. In the Northwestern Mediterranean Sea, the Northern Current (Fernández et al., 2005) could be considered as a dispersal mechanism for A. antennatus. Thus, blue and red shrimp larval stages have been found in the upper water layers (Carreton et al., 2019 and references therein), which means that they are likely dispersed by this surface current. Furthermore, juvenile and/or adult migration may contribute to genetic connectivity. Indeed, it has been proven that adults have the capability of movement, as they do seasonal displacements between the open slope and submarine canyons, mainly related to the spawning period (Sardà et al., 1997, 2004). Females and males could present a different degree of dispersal, as several genetic studies using microsatellite loci have indicated. Planella et al. (2019) found that Palamós’ females mated with both males from the same location and males from other fishing grounds, suggesting a migration of A. antennatus’ adult males from one location to another. Recently, Abras et al. (2021) detected significant juvenile males’ incomes at the Palamós’ fishing ground from neighbouring ones.
On the other hand, the temporal analyses showed that the regional patterns of population genetic structure remain stable for females not only between years (Table 3 and Supplementary Table S3) but also between seasons within years (Table 5) despite changes in the relative abundance of cohorts during winter and summer collections (Carbonell et al., 1999). By contrast, a small but significant differentiation was found between 2016 and 2017 winter collections, when sex ratios were close to 1:1 (Agulló et al., 2020). Thus, specific patterns of males’ migration beyond passive larval dispersal could be the main reason for the temporal differentiation found in Agulló et al. (2020) at some of the studied fishing grounds. Cannas et al. (2012) had already pointed out a sex-biassed dispersal favouring of A. antennatus females in the Central Mediterranean Sea.
Management implications
Fisheries management must take into account the conservation of genetic diversity to ensure the perpetuation of natural populations, avoiding their overexploitation (Waples & Naish, 2009). Since variations in the genetic population structure, including a decrease in genetic variability, can occur in a short period of time, a temporal monitoring is recommended; both at levels of genetic diversity within the population and at the levels of connectivity, to study long-term changes (Carvalho & Hauser, 1994; Allendorf et al., 2008).
The effective population size (Ne) is a crucial parameter to draw inferences on the adaptive potential of the population and its value should not be lower than 1000 to ensure population sustainability and evolutionary potential, thus minimizing extinction risk (Frankham et al., 2014). A. antennatus fishing grounds in the GSA6 displayed moderate and high Ne values (Table 2), which means that the blue and red shrimp fishery would maintain the genetic variability in the long term.
On the other hand, even though our temporal analysis includes four sampling campaigns in two years, 2016 and 2017, more than 2000 female individuals were genotyped. The genetic information obtained is a forceful estimation of the genetic connectivity and the temporal stability of the blue and red shrimp populations in the GSA6.
The low and non-significant differentiation detected among females from distant A. antennatus fishing grounds indicates a high gene flow that would limit local adaptation (Hauser & Carvalho, 2008). Therefore, we advise considering A. antennatus in the GSA6 as a single management unit, although the boundaries could be expanded beyond the limits of GSA6 if genetic homogeneity was detected in adjacent Mediterranean Subareas. Nevertheless, because of genetic instability indicated at some fishing grounds between consecutive winter samples by Agulló et al (2020), when males are present (Carbonell et al., 1999), and females show increased dispersal (Cannas et al., 2012), keeping each fishing ground under local monitoring is needed for a sustainable exploitation of A. antennatus. Thus, the establishment of a fishery closure for the blue and red shrimp during winter in the GSA6 as declared for this and other Spanish demersal fisheries (Boletín Oficial del Estado, 2020) is likely to improve male recruitment and reproduction in the next spawning season. Finally, a future management plan of the blue and red shrimp fishery integrating genetic data, biological information and hydrodynamic modelling would allow the sustainable exploitation of the fishing grounds (Ward, 2000; Reiss et al., 2009).
Data and material availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Abras, A., J. L. García-Marín, S. Heras, M. Vera, M. Agulló, L. Planella & M. I. Roldán, 2021. Male deep-sea shrimps Aristeus antennatus at fishing grounds: growth and first evaluation of recruitment by multilocus genotyping. Life 11: 116.
Addison, J. A. & M. W. Hart, 2005. Spawning, copulation and inbreeding coefficients in marine invertebrates. Biology Letters 1: 450–453.
Agulló, M., S. Heras, J. L. García-Marín, M. Vera, L. Planella & M. I. Roldán, 2020. Genetic analyses reveal temporal stability and connectivity pattern in blue and red shrimp Aristeus antennatus populations. Scientific Reports 10: 21505.
Allendorf, F. W., P. R. England, G. Luikart, P. A. Ritchie & N. Ryman, 2008. Genetic effects of harvest on wild animal populations. Trends in Ecology & Evolution 23: 327–337.
Boletín Oficial del Estado, 2020. Orden APA/1212/2020, de 16 de diciembre, por la que se establecen zonas de veda espaciotemporal para la modalidad de arrastre de fondo y cerco en determinadas zonas del litoral mediterráneo para el periodo 2021–2022. BOE 331: 117341–117346.
Cannas, R., F. Sacco, M. C. Follesa, A. Sabatini, M. Arculeo, S. Lo Brutto, T. Maggio, A. M. Deiana & A. Cau, 2012. Genetic variability of the blue and red shrimp Aristeus antennatus in the Western Mediterranean Sea inferred by DNA microsatellite loci. Marine Ecology 33: 350–363.
Carbonell, A., M. Carbonell, M. Demestre, A. Grau & S. Monserrat, 1999. The red shrimp Aristeus antennatus (Risso, 1816) fishery and biology in the Balearic Islands, Western Mediterranean. Fisheries Research 44: 1–13.
Carbonell, A., 2005. Evaluación de la gamba rosada, Aristeus antennatus (Risso 1816), en el Mar Balear. PhD dissertation, University of the Balearic Islands, Mallorca, Spain: 212 pp.
Carbonell, A., J. Lloret & M. Demestre, 2008. Relationship between condition and recruitment success of red shrimp (Aristeus antennatus) in the Balearic Sea (Northwestern Mediterranean). Journal of Marine Systems 71: 403–412.
Carretón, M., J. B. Company, L. Planella, S. Heras, J. L. García-Marín, M. Agulló, M. Clavel-Henry, G. Rotllant, A. Dos Santos & M. I. Roldán, 2019. Morphological identification and molecular confirmation of the deep-sea blue and red shrimp Aristeus antennatus larvae. PeerJ 7: e6063.
Carvalho, G. R. & L. Hauser, 1994. Molecular genetics and the stock concept in fisheries. Reviews in Fish Biology and Fisheries 4: 326–350.
Chapuis, M. P. & A. Estoup, 2007. Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution 24: 621–631.
Demestre, M. & J. M. Fortuño, 1992. Reproduction of the deep-water shrimp Aristeus antennatus (Decapoda: Dendrobranchiata). Marine Ecology Progress Series 84: 41–51.
Demestre, M. & P. Martín, 1993. Optimum exploitation of a demersal resource in the western Mediterranean: the fishery of the deep-water shrimp Aristeus antennatus (Risso, 1816). Scientia Marina 57: 175–182.
Demestre, M., 1995. Moult activity-related spawning success in the Mediterranean deep-water shrimp Aristeus antennatus (Decapoda: Dendrobranchiata). Marine Ecology Progress Series 127: 57–64.
Deval, M. C. & K. Kapiris, 2016. A review of biological patterns of the blue-red shrimp Aristeus antennatus in the Mediterranean Sea: a case study of the population of Antalya Bay, eastern Mediterranean Sea. Scientia Marina 80: 339–348.
Do, C., R. S. Waples, D. Peel, G. M. Macbeth, B. J. Tillett & J. R. Ovenden, 2014. NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources 14: 209–214.
Esteban, A., 2018. Stock Assessment Form Demersal species Reference year 2017. Reporting year 2018, FAO, Rome: 32.
European Council, 2002. Council Regulation (EC) No 2371/2002 of 20 December 2002 on the conservation and sustainable exploitation of fisheries resources under the Common Fisheries Policy. Official Journal of the European Communities L 358: 59–80.
European Council, 2019. Regulation (EU) 2019/1022 of the European Parliament and of the Council of 20 June 2019 establishing a multiannual plan for the fisheries exploiting demersal stocks in the western Mediterranean Sea and amending Regulation (EU) No 508/2014. Official Journal of the European Union L 172: 1–17.
Excoffier, L., P. E. Smouse & J. M. Quattro, 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Excoffier, L. & H. E. L. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567.
FAO, 2006. General Fisheries Commission for the Mediterranean. Report of the ninth session of the Scientific Advisory Committee. FAO Fisheries Report 814, Rome, Italy: 106 pp.
Fernández, M. V., S. Heras, F. Maltagliati, A. Turco & M. I. Roldán, 2011. Genetic structure in the blue and red shrimp Aristeus antennatus and the role played by hydrographical and oceanographical barriers. Marine Ecology Progress Series 421: 163–171.
Fernández, M. V., S. Heras, F. Maltagliati & M. I. Roldán, 2013. Deep genetic divergence in giant red shrimp Aristaeomorpha foliacea (Risso, 1827) across a wide distributional range. Journal of Sea Research 76: 146–153.
Fernández, V., D. E. Dietrich, R. L. Haney & J. Tintoré, 2005. Mesoscale, seasonal and interannual variability in the Mediterranean Sea using a numerical ocean model. Progress in Oceanography 66: 321–340.
Frankham, R., C. J. A. Bradshaw & B. W. Brook, 2014. Genetics in conservation management: revised recommendations for the 50/500 rules, red list criteria and population viability analyses. Biological Conservation 170: 56–63.
Gorelli, G., J. B. Company & F. Sardà, 2014. Management strategies for the fishery of the red shrimp Aristeus antennatus in Catalonia (NE Spain). Marine Stewardship Council Science Series 2: 116–127.
Goudet, J., 2001. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Accessed 15 Sep 2020. https://www2.unil.ch/popgen/softwares/fstat.htm.
Hauser, L. & G. R. Carvalho, 2008. Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish and Fisheries 9: 333–362.
Heras, S., L. Planella, I. Caldarazzo, M. Vera, J. L. García-Marín & M. I. Roldán, 2016. Development and characterization of novel microsatellite markers by next generation sequencing for the blue and red shrimp Aristeus antennatus. PeerJ 4: e2200.
Heras, S., L. Planella, J. L. García-Marín, M. Vera & M. I. Roldán, 2019. Genetic structure and population connectivity of the blue and red shrimp Aristeus antennatus. Scientific Reports 9: 13531.
Hill, W. G., 1981. Estimation of effective population size from data on linkage disequilibrium. Genetics Research 38: 209–216.
Leitão, F., 2019. Mean size of the landed catch: a fishery community index for trend assessment in exploited marine ecosystems. Frontiers in Marine Science 6: 302.
Maggio, T., S. Lo Brutto, R. Cannas, A. M. Deiana & M. Arculeo, 2009. Environmental features of deep-sea habitats linked to the genetic population structure of a crustacean species in the Mediterranean Sea. Marine Ecology 30: 354–365.
Marra, A., S. Mona, R. M. Sà, G. D’Onghia & P. Maiorano, 2015. Population genetic history of Aristeus antennatus (Crustacea: Decapoda) in the Western and Central Mediterranean Sea. PLoS ONE 10: e0117272.
Massutí, E., S. Monserrat, P. Oliver, J. Moranta, J. L. López-Jurado, M. Marcos, M. Hidalgo, B. Guijarro, A. Carbonell & P. Pereda, 2008. The influence of oceanographic scenarios on the population dynamics of demersal resources in the western Mediterranean; Hypothesis for hake and red shrimp off Balearic Islands. Journal of Marine Systems 71: 421–438.
Maynou, F., 2008. Environmental causes of the fluctuations of red shrimp (Aristeus antennatus) landings in the Catalan Sea. Journal of Marine Systems 71: 294–302.
Nanami, A., T. Sato, Y. Kawabata & J. Okuyama, 2017. Spawning aggregation of white-streaked grouper Epinephelus ongus: spatial distribution and annual variation in the fish density within a spawning ground. PeerJ 5: e3000.
Pilling, G. M., L. T. Kell, T. Hutton, P. J. Bromley, A. N. Tidd & L. J. Bolle, 2008. Can economic and biological management objectives be achieved by the use of MSY-based reference points? A North Sea plaice (Pleuronectes platessa) and sole (Solea solea) case study. ICES Journal of Marine Science 65: 1069–1080.
Planella, L., M. Vera, J. L. García-Marín, S. Heras & M. I. Roldán, 2019. Mating structure of the blue and red shrimp, Aristeus antennatus (Risso, 1816) characterized by relatedness analysis. Scientific Reports 9: 7227.
Rahman, M. M. & J. Ohtomi, 2017. Reproductive biology of the deep-water velvet shrimp Metapenaeopsis sibogae (De Man, 1907) (Decapoda: Penaeidae). Journal of Crustacean Biology 37: 743–752.
Reiss, H., G. Hoarau, M. Dickey-Collas & W. J. Wolff, 2009. Genetic population structure of marine fish: mismatch between biological and fisheries management units. Fish and Fisheries 10: 361–395.
Rice, W. R., 1989. Analyzing tables of statistical tests. Evolution 43: 223–225.
Rohlf, F. J., 1993. NTSYS-pc. Numerical Taxonomy and Multivariate Analysis System, Version 2.1, Setauket, New York: 43 pp.
Roldán, M. I., S. Heras, R. Patellani & F. Maltagliati, 2009. Analysis of genetic structure of the red shrimp Aristeus antennatus from the Western Mediterranean employing two mitochondrial regions. Genetica 136: 1–4.
Roldán, M. I., L. Planella, S. Heras & M. V. Fernández, 2014. Genetic analyses of two spawning stocks of the short-finned squid (Illex argentinus) using nuclear and mitochondrial data. Comptes Rendus Biologies 337: 503–512.
Rousset, F., 1997. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Rousset, F., 2008. GENEPOP´007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources 8: 103–106.
Ryman, N. & S. Palm, 2006. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Molecular Ecology Notes 6: 600–602.
Sardà, F., F. Maynou & L. Talló, 1997. Seasonal and spatial mobility patterns of rose shrimp Aristeus antennatus in the Western Mediterranean: results of a long-term study. Marine Ecology Progress Series 159: 133–141.
Sardà, F., C. Bas, M. I. Roldán, C. Pla & J. Lleonart, 1998. Enzymatic and morphometric analyses in mediterranean populations of the rose shrimp, Aristeus antennatus (Risso, 1816). Journal of Experimental Marine Biology and Ecology 221: 131–144.
Sardà, F., J. B. Company & F. Maynou, 2003. Deep-sea shrimp Aristeus antennatus Risso 1816 in the Catalan Sea, a review and perspectives. Journal of Northwest Atlantic Fishery Science 31: 127–136.
Sardà, F., G. D’Onghia, C. Y. Politou, J. B. Company, P. Maiorano & K. Kapiris, 2004. Deep-sea distribution, biological and ecological aspects of Aristeus antennatus (Risso, 1816) in the western and central Mediterranean Sea. Scientia Marina 68: 117–127.
Sardà, F., M. I. Roldán, S. Heras & F. Maltagliati, 2010. Influence of the genetic structure of the red and blue shrimp, Aristeus antennatus (Risso, 1816), on the sustainability of a deep-sea population along a depth gradient in the western Mediterranean. Scientia Marina 74: 569–575.
Sundqvist, L., K. Keenan, M. Zackrisson, P. Prodöhl & D. Kleinhans, 2016. Directional genetic differentiation and relative migration. Ecology and Evolution 6: 3461–3475.
Van Oosterhout, C., W. F. Hutchinson, D. P. M. Wills & P. Shipley, 2004. Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4: 535–538.
Waples, R. S., 1998. Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. Journal of Heredity 89: 438–450.
Waples, R. S., 2006. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conservation Genetics 7: 167–184.
Waples, R. S., A. E. Punt & J. M. Cope, 2008. Integrating genetic data into management of marine resources: how can we do it better? Fish and Fisheries 9: 423–449.
Waples, R. S. & K. A. Naish, 2009. Genetic and evolutionary considerations in fishery management: research needs for the future. In Beamish, R. J. & B. J. Rothschild (eds), The Future of Fisheries Science in North America, Fish & Fisheries Series Springer, Dordrecht: 427–451.
Ward, R. D., 2000. Genetics in fisheries management. Hydrobiologia 420: 191–201.
Weir, B. S. & C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Acknowledgements
We gratefully thank the ship-owners Francisco Torné (Eli-Hermi II), Manel Noguera (Port de Roses), Conrad Masseguer (Nova Gasela), Pere Pérez (Peret II), Marc Pons (Avi Salvador), Rafael Montoya (Avi Pau), Juan Antonio Sepulcre (La androna) and Jorge Castejón (Isabel y Andrés) for assistance in sampling collection. This paper was considerably improved by suggestions from two anonymous reviewers and the Associate Editor.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was supported by a grant from Spanish Ministerio de Economia y Competitividad (CTM2014-54648-C2-2-R) to MIR. LP and MA benefited from predoctoral fellowship from the Universitat de Girona (BR2014 and IFUdG2018, respectively).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Ethical approval
Not applicable.
Additional information
Handling Editor: Cécile Fauvelot
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Agulló, M., Heras, S., García-Marín, JL. et al. An evaluation of the genetic connectivity and temporal stability of the blue and red shrimp Aristeus antennatus: a case study of spawning females’ grounds in the Western Mediterranean Sea. Hydrobiologia 849, 2043–2055 (2022). https://doi.org/10.1007/s10750-022-04847-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-022-04847-3