
Abstract
A high number of grayling (Thymallus) species have been described from the Altai-Sayan mountain region, for which little to no genetic information is available. We investigated genetic relationships within this genus of salmonid fishes using mtDNA and microsatellite markers. The analysis focused on three putative species, Markakol grayling (T. brevicephalus), Upper Ob grayling (T. nikolskyi) and Mongolian grayling (T. brevirostris). We integrated these data with mtDNA sequences from eight other grayling species, including two of geographic proximity to the study area. Phylogenetic results revealed three pairs of reciprocally monophyletic sister species, two of which were phylogenetically juxtaposed across isolated drainage systems. Based on microsatellite analysis (up to 10 loci) no evidence of hybridization or introgression was found among species, supporting the mtDNA phylogeny. Based on a time-calibrated tree, divergence times between the focal taxa ranged from 0.36 to 1.1 MY. The genetic data support the distinction of these species and underscore the importance of paleohydrological dynamics in this biogeographically complex region. Well-documented mega-flood events in the region provide a model of how the contemporary cross-basin distribution of these species may have evolved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Paleoclimatic processes have played a major role in shaping species distributions and diversity (Wiens & Donoghue, 2004). A better understanding of the legacies of such processes is crucial for conserving biodiversity and modeling predictions of how species, as well as ecosystems, will respond to anthropogenic climate change (Svenning et al., 2015). The interplay between climatic processes and evolution may be markedly different for freshwater compared to terrestrial organisms, as drainage position and historical connectivity play a larger role in explaining both species ranges and phylogeographic structure (Dias et al., 2014; Carvajal-Quintero et al., 2019). The generalization that paleohydrological dynamics and historical connectivity can strongly influence contemporary species diversity and distributions is supported by many studies of freshwater fishes in North America (Griffiths, 2015; Matamoros et al., 2016), Europe (Buj et al., 2017; Levin et al., 2019), New Zealand (Craw et al., 2007), Australia (Waters et al., 2019), Asia (Froufe et al., 2003, 2008; de Bruyn et al., 2013) and South America (Schönhuth et al., 2011). The origins of this conceptual development began with a large number of phylogeographic studies that attempted to link phylogeographic patterns with contemporary drainage networks and ocean basins. In Europe, such studies on freshwater fishes began with brown trout Salmo trutta Linnaeus, 1758 (Bernatchez et al., 1992), European chub Squalius cephalus (Linneaus, 1758) (Durand et al., 1999), European perch Perca fluviatilis Linnaeus, 1758 (Nesbo et al., 1999) and European grayling Thymallus thymallus (Linnaeus, 1758) (Weiss et al., 2002).
Similar studies were carried out throughout Eurasia, including Siberia (Stepien et al., 1998; Froufe et al., 2003). Thymallus spp. (graylings) were the target of one of the first broad-scale phylogeographic analyses of freshwater fishes in Siberia (Koskinen et al., 2002). While the general phylogeographic patterns uncovered in that study largely reflected contemporary drainage networks, results also agreed with a cataclysmic paleohydrological event involving ancient Lake Baikal. Subsequent studies on the diversity and distribution of graylings in Siberia took advantage of the growing knowledge of large-scale river network dynamics in this area, associated with Quaternary climatic oscillations and glacial cycles. While the paleogeological history of Lake Baikal has attracted considerable research interest, elsewhere in Siberia the potential effects of Pleistocene glaciations on the hydrological network were underestimated or understudied. However, beginning with the controversial perspectives of Grosswald (1998), there has been an increased understanding of the large scope and magnitude of the paleohydrological dynamics of Siberian rivers (e.g. Komatsu et al., 2016; Margold et al., 2018).
Thus far, salmonid fishes, including Thymallus, have proved to be good models for investigating the interplay between such large-scale paleohydrological processes and biological diversification (Koskinen et al., 2002; Froufe et al., 2005; Weiss et al., 2006, 2007; Froufe et al., 2008; Weiss et al., 2020). These studies highlight the shifting phases of hydrological isolation and historical connectivity within and between major river drainage networks such as the Amur, Lena, Enisei and Ob. These paleohydrological insights are congruent with the description of specific events (e.g. mega-floods) that have helped shape the distributions of freshwater organisms (e.g. Ivanov et al., 2016; Komatsu et al., 2016; Arzhannikov et al., 2018).
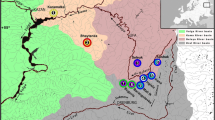
Nevertheless, there is still much uncertainty concerning the biological consequences of paleohydrological dynamics, especially where knowledge of faunal diversity and distributions is incomplete. The Altai-Sayan mountain region, in particular, has been insufficiently investigated concerning the distribution and systematics of its aquatic fauna, including grayling. Here, the headwaters of the Ob and Enisei river drainages are juxtaposed with the endorheic basin of the Khovd and Zavkahn river drainages (Fig. 1). These rivers harbor several putative grayling species with unclear evolutionary relationships (i.e. T. baicalensis Dybowski, 1874; T. brevirostris Kessler, 1879; T. nikolskyi Kaschenko, 1899; T. nigrescens Dorogostaisky, 1923; T. brevicephalus Mitrofanov, 1971 and T. svetovidovi Knizhin & Weiss, 2009). Thymallus nigrescens is endemic to Lake Chuvsgol, located in the upper Enisei River drainage in Mongolia, whereas T. svetovidovi is found in another branch of the Enisei drainage (Knizhin & Weiss, 2009) that is geographically close to Lake Chuvsgol, but in terms of the contemporary hydrological network, very distant. Thymallus nikolskyi, first described over 100 years ago (Kaschenko, 1899) is reported from several tributaries of the adjacent upper Ob River drainage (Romanov et al., 2016; Dyldin et al., 2017), whereas T. brevicephalus is found in the nearby Irtysh River branch of the Ob River drainage, in Lake Markakol (Mitronfanov, 1971) as well as in adjacent tributaries flowing from the Altai Mountains to the Irtysh. Thymallus baicalensis is the commonly occurring grayling inhabiting most of the Enisei River drainage including both Lake Baikal and its Selenga River headwaters in northern Mongolia (see Weiss et al., 2007). Problematic for the understanding of these species is that they are often reported as Arctic grayling (T. arcticus Pallas, 1776) or assigned to subspecies of Arctic grayling (see Knizhin et al., 2006a and the review of Dyldin et al., 2017). Thymallus brevirostris occurs in the endorheic basin of the Khovd and Zavkahn river drainages of Western Mongolia and the Tuva Republic in Russia. The phenotypic variability of T. brevirostris in Western Mongolia is large, and not restricted to the large-piscivorous phenotype most commonly associated with the taxon (Knizhin et al., 2008; Slynko et al., 2010).

a Overview map of Asia showing the location of the study area in a box, and the flow of three major Siberian river systems; Altai-Sayan mountain region in grey; b overview of the upper Ob and Enisei River drainages as well as the adjacent endorheic basin of Western Mongolia, including the focal area of this study. The numbered sample sites are color-coded to represent the three grayling species known to occur in this region; green = T. brevirostris, red = T. brevicephalus, and blue = T. nikolskyi (same as in Fig. 3). Shown are the approximate distribution ranges of these species along with those of T. svetovidovi, T. baicalensis and T. nigrescens as well as several hypothesized mega-flood events shown with black arrows. These events are (a) Tunguska–Enisei shortcut; (b) Altai as shown in Komatsu et al., 2016); (c) Mogen–Buren valley outburst (Agatova et al., 2015); (d) Sayan flood (Komatsu et al., 2016); and (e) Angara breakout (Arzhannikov et al., 2018)
For a better understanding of distribution patterns of freshwater fishes, it is essential to integrate paleohydrological and phylogenetic information (Carvajal-Quintero et al., 2019). Systematic studies of the region’s freshwater fauna will provide not only a better understanding of the evolutionary history of specific organisms but also potentially help fill knowledge gaps in the paleohydrological history itself (Koskinen et al., 2002; Wong et al., 2004; Near & Keck, 2005). The extremely dynamic paleohydrological events in the Altai-Sayan mountain region were asynchronous with those in Siberia and western Central Asia (Gillespie et al., 2008). These events involved ice-dammed lakes and fluvial catastrophes, exceeding in magnitude the renowned flood of Lake Missoula in the Western USA (Agatova & Nepop, 2019 and citations therein). Mega-floods are well documented in the Lena (Margold et al., 2018), Enisei (Arzhannikov et al., 2018) and Ob river drainages (Bohorquez et al., 2015; Komatsu et al., 2016). The latter has been estimated to be two orders of magnitude larger than contemporary floods of the world’s largest rivers (Agatova & Nepop, 2019 and citations therein). While these events were probably largely catastrophic for many organisms in the short-term, they also likely resulted in drainage rearrangements and cross-drainage colonization corridors that led to lineage distributions that contrast with the freshwater phylogeographic paradigm of isolation among drainages (Avise, 2007; Lerceteau-Köhler et al., 2013).
Here we constructed an mtDNA molecular phylogeny of putative (following Dyldin et al., 2017 and Frick et al., 2020) Thymallus species occurring in the Altai-Sayan region, using both original data and data obtained from the literature (Koskinen et al., 2002; Froufe et al., 2005; Knizhin et al., 2008; Knizhin & Weiss, 2009). Using mitochondrial and microsatellite data, we further investigated the molecular phylogeographic and population genetic structure of those three taxa, distributed in the Irtysh and Biya sub-drainages of the Ob River drainage, and the endorheic basin of Western Mongolia. We also used coalescent modeling to help understand the recent evolutionary history of T. brevicephalus. For these analyses, we used both mitochondrial and microsatellite markers. No population genetic data are presently available for these three taxa and, for T. brevicephalus, this represents the first investigation with molecular genetic markers.
Materials and methods
In 2012, 89 grayling were captured via gill nets from three locations in the upper Irtysh drainage in Kazachstan: the Kara-Kaba River (Kka); the Urunkhaika River (Rur), a tributary of Lake Markakol; and the Kaldzhir River (Kal), the lake’s primary outflow (Fig. 1; Table 1). Additional samples (N = 40) from Lake Teletskoye (Biy) in the upper Ob River drainage, and various locations in the Khovd (Khg, Kht, Khv, Tol, Ach), and Zavkhan (Kn) river drainages (N = 62) of Western Mongolia were included (Knizhin et al., 2008; Fig. 1; Table 1). Whole genomic DNA was isolated from ethanol preserved fin clips using a high salt (ammonium acetate) extraction protocol (Sambrook et al., 1989). The complete mtDNA control region (CR) together with partial segments of the two flanking tRNA (Proline & Phenylalanine) genes was amplified in 32 samples from the Irtysh River drainage basin using the primers CRRII_Int2F (5′-GGA ATC CCC CGG CTT CTA C-3′), CRI_ Int1R (5′-ACT TCC TGG TTT AGG GGT TTG AC-3′) and the internal primer Int5R (5′-ATA TAA GAG AAC GCC CGG CT-3′). Additionally, six samples from Lake Teletskoye were amplified with the primers LRBT-25 and LRBT-1195 (Uiblein et al., 2001). Use of the mtDNA CR allows comparison with a large number of publicly available sequences, and the gene has proven useful for both within- and between-species analyses in the genus (Weiss et al., 2002; Koskinen et al., 2002; Froufe et al., 2005; Weiss et al., 2020). PCRs were done in 25 µl with each reaction containing 13.5 µl H2O, 5 µl of Phusion GC Buffer (Thermo Scientific), 0.5 µl of 10 mM dNTPs, 1.25 µl 10 mM of each primer, 2.5 µl Phusion Polymerase, and 1 µl of 100 ng/µl DNA. Initial PCR denaturation was at 98°C for 30 s, followed by 35 cycles at 98°C for 10 s, annealing at 57°C (LRBT-25 and LRBT-1195) or 55°C (CRII_Int2F, CRI_Int1R and Int5R) for 30 s and 72°C for 30 s and final extension at 72°C for 10 min. PCR products were purified with ExoSap-IT (Amersham Biosciences) and sequenced on an ABI 3130xl Genetic Analyzer using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Phylogenetic analysis
New sequences were aligned together with additional CR sequences (N = 66) from previously published studies (see Table 1 and Supplementary Material S1). Including two outgroup taxa, a total of 104 sequences were aligned using the Mafft multiple sequence global pair alignment (Katoh & Stanley, 2013) and reduced to unique haplotypes with the DNA collapser tool on the FaBox 1.5 platform (Villeseon, 2007). Phylogenetic analyses were carried out with Maximum Likelihood (ML) and Bayesian Inference (BI). The best-fit model (HKY+I+G) of nucleotide substitution was identified using jModelTest2 v2.1.6 (Darriba et al., 2012). The ML phylogeny was reconstructed using RAxML-HPC2, v8.2.12 (Stamatakis, 2014), with the model GTR + G (following RAxML authors’ recommendation) and 1000 bootstrap iterations. Both jModelTest and RAxML were run in the CIPRES gateway (Miller et al., 2010). The BI analysis was performed with MrBayes v3.2.6 (Ronquist & Huelsenbeck, 2003) with the model HKY+I+G, again run in the CIPRES gateway. Two independent runs of 2 × 107 generations, sampled every 2000 generations, were performed, each with four chains (three hot, one cold). A burn-in of 20,000 generations was determined by assessing ESS values with Tracer v1.7.1 (Rambaut et al., 2018). The two independent runs were merged, and a 50% majority rule tree generated with MrBayes v3.2.6 offline. To gain a non-bifurcating perspective of haplotype relationships among drainages, a haplotype network was generated with a 95% parsimony criterion (Templeton et al., 1992) using TCS v1.13 (Clement et al., 2000). For this analysis, indels were coded as a fifth state character.
To estimate divergence times between species, a time-calibrated phylogeny was produced in BEAST (Drummond & Rambaut, 2007; Suchard & Rambaut, 2009) in CIPRES science gateway (Miller et al., 2010). The analysis was carried out with the same substitution model used for the MrBayes phylogeny and uncorrelated relaxed molecular clock priors with lognormal distributions (Drummond et al., 2006). The Birth–Death speciation process (Gernhard, 2008) was chosen as a tree prior, given the dataset comprised intra- and inter-specific relationships. The substitution rate prior was set with a normal distribution and 1% per MY (average 0.01 and 0.002 SD) following the mtDNA molecule calibration for salmonids (Smith, 1992) and the reported lower bound of the expansion described in Koskinen et al. (2002). Analysis was performed with one 30 M MCMC iterations run, sampling every 3000 runs. Burn-in and run convergence (ESS > 200) were determined using Tracer v1.7.1 (Rambaut et al., 2018). The final tree was produced in TreeAnnotator v1.10.4 (in the BEAST package) and formatted in FigTree v1.4.4 (Rambaut, 2012).
To characterize further the differences within and among putative species, the nucleotide diversity (Nei & Li, 1979) was calculated using DnaSP v5 (Librado & Rozas, 2009). Net nucleotide divergence (Dα) between putative species (uncorrected p-distances) was calculated using MEGA v10.0.05 (Kumar et al., 2018).
Population-level genetic structure
To evaluate genetic relationships within and among sample locations and provide bi-parentally inherited nuclear DNA markers to compliment the mtDNA-based phylogenetic reconstruction, we examined allelic variation across 10 microsatellite loci. However, due to the high genetic divergence among groups of samples, presumably spanning different species, not all loci amplified or revealed polymorphism for all samples. Thus, all 10 loci were applied to samples from the upper Irtysh drainage (T. brevicephalus), while only seven loci could be applied to samples from the Biya River (T. nikolskyi) and Western Mongolia (T. brevirostris). Loci were amplified using duplex or triplex PCRs following conditions in Weiss et al. (2013) (see Table 2) on an ABI 3130xl Genetic Analyzer and scored using Gene Mapper software v3.7 (Applied Biosystems). The number of alleles per locus, deviations from Hardy–Weinberg Equilibrium (HWE) based on FIS values, and deviations from linkage equilibrium (LE) were calculated with FSTAT v2.9.3.2 (Goudet, 2001). Observed and expected heterozygosity, and two different pairwise measures of differentiation, one based on an infinite allele model (an FST analog) and one based on a stepwise mutation model (RST), were calculated using ARLEQUIN v3.5.1.2 (Excoffier & Lischer, 2010). The estimates of pairwise differentiation were used to evaluate the potential contrast between differentiation within and between putative species across multiple sampling locations.
Genetic relationships among individual genotypes of Thymallus spp. (without regard to location or assumed taxonomic assignment) were graphically represented using a factorial correspondence analysis (FCA) computed in GENETIX v4.05.2 (Belkhir et al., 1996–2004). This multi-variate procedure is based on a matrix of binary input variables representing the presence or absence of each microsatellite allele across all loci. Additionally, a Bayesian clustering method in STRUCTURE v2.3.4 (Pritchard et al., 2000) was used to assess the overall genetic structure in the data, again, without a-prior assignment of individuals to species or drainages. The posterior probabilities of K (number of populations) were estimated assuming uniform prior values of K between 1 and 6. Structure was run for 100,000 iterations, of which the first 50,000 iterations were discarded as burn-in and five independent replicates of the MCMC were conducted for each value of K assuming an admixture model and correlated allele frequencies. We determined the numbers of K that best fit the data with the ad-hoc Delta K statistic (Evanno et al., 2005) using the online tool Structure Harvester (Earl & vonHoldt, 2012). Graphs of individual Q values (estimated percent ancestry) were displayed to evaluate potential admixture between sample locations.
Thymallus nikloskyi DIY-ABC modeling
Preliminary analysis showed significant divergence between the two sample locations of T. nikolskyi directly connected to Lake Markakol (Rur and Kal) and the sample location in the more distant Kara-Kaba River (Kka). Thus, we decided to estimate divergence times and population history of these three population samples using a coalescent-based Approximate Bayesian Computation (ABC) algorithm in the program package DIY-ABC v2.0.4 (Cornuet et al., 2014). Demographic parameters were simulated for three hypothetical evolutionary scenarios (Fig. 2) and then compared to the observed real data to rank those scenarios (Csilléry et al., 2010). The parameters of interest were the coalescence time (in generations) since population divergence, discrete change in effective population size, and the time (and rate) of population admixture. We used a default uniform distribution and broad priors for all parameters. Effective population sizes were set to range from 30 to 40,000 for all populations and events. Priors for coalescence times (in generations) were set as follows: t1 = the most recent split between populations ranging from 1 to 10,000; t12 = time of population size change ranging from 30 to 20,000, whereby t12 ≥ t1; and t2 = the initial split from an ancestral population ranging from 50 to 40,000 whereby t2 > t1, t2 > t12. For scenario 2, an admixture event was modeled at time t1, with the Rur population resulting from admixture between Kka and Kal, with an admixture rate of ra ranging from 0.001 to 0.999. An equal prior probability was assumed for each competing scenario.

Graphic representation of the three demographic scenarios (1, 2, and 3) that were modeled for the three population samples of T. brevicephalus, using DIY-ABC, and the comparative posterior probabilities of each model fitting the real data. The demographic modeling used both 10 microsatellite loci and the mtDNA CR sequences. See “Materials and methods” for details
To assign mutation rates and evolutionary models, genetic loci were divided into three groups: dinucleotide microsatellites, tetranucleotide microsatellites, and the mtDNA CR. Microsatellites had a possible range of 40 contiguous allelic states. The 10 loci were assumed to follow a Generalized Stepwise Mutation model (Estoup et al., 2002), where each mutation involves an increase or decrease in allele length by one or more several repeat motifs. To apply this model, the parameters mean mutation rate (µ) and a geometric distribution (P) from which the number of repeats is drawn, are required. The mean mutation rates for both sets of microsatellite loci were assumed to follow a uniform distribution with the prior rate per locus per generation set to range from 10−6 to 9 × 10−4 for the dinucleotide loci, and from 10−4 to 9 × 10−4 for the tetranucleotide loci. The geometric distribution parameter P was set to range from 0.1 to 0.3. Additionally, each microsatellite locus was characterized by an individual mutation rate µi, drawn from an individual geometric distribution Pi. The mtDNA CR was assumed to follow the Hasegawa-Kishino-Yano substitution model (Hasegawa et al., 1985) with a Gamma shape parameter of 0.596, and an assumption of invariable sites set at 61%. The mean mutation rate per site per generation for the mtDNA locus was also assumed to be uniform with a prior set from 10−9 to 10−7 per site per generation. We simulated 106 datasets for each explored scenario. Posterior probabilities of each scenario were compared using local logistic regression on 1% of the closest simulated data set.
Results
Phylogenetic analysis
The final alignment encompassed 1009 bp of the CR, 68 bp of the tRNA proline gene and 10 bp of the tRNA phenylalanine gene. There were 161 variable sites, 129 of which were parsimony informative. BI and ML methods resulted in topologies that were congruent for the focal taxa, so only the BI tree is shown (see Supplementary Material S2 for the ML topology), with node support values depicted from both approaches (Fig. 3). Incongruences in non-focal taxa were related to the placement of branches with very low support in both trees. Net mean divergence (uncorrected p-distances) between species ranged from a minimum of 0.2% (T. baicalensis Dybowski, 1874 vs. T. nigrescens Dorogostaisky, 1923) to a maximum of 5.6% (T. tugarinae Knizhin, Antonov, Safronov & Weiss, 2007 vs. T. grubii) (Fig. 3; Table 3). After the splits involving Amur River drainage species (T. tugarinae, T. grubii, T. flavomaculatus, T. burejensis Antonov, 2004), T. thymallus, T. arcticus and T. baicalolenensis Matveev, Samusenok, Pronin & Tel’pukhovsky, 2005, three monophyletic clades remained, representing grayling of the Altai-Sayan mountain region. Clade A contains samples from T. nikolskyi in the upper Ob River drainage and T. svetovidovi in the upper Enisei River drainage. Clade B contains haplotypes from T. nigrescens and T. baicalensis in the Lake Baikal branch of the Enisei drainage. Clade C includes all haplotypes from all samples of T. brevirostris from the endorheic basin of Western Mongolia as well as T. brevicephalus from the upper Irtysh River drainage. The monophyly of T. brevirostris and T. brevicephalus (Clade C) are each supported by moderate ML bootstrap values (69–85%) and high BI values (> 95%). The net mean divergence between these two species was 0.4%, between T. nikolskyi and T. brevirostris 1.5%, and between T. nikolskyi and T. brevicepahlus 1.7%. T. nikolskyi showed more nucleotide diversity (π = 0.0032) than T. brevirostris (π = 0.0020) and T. brevicephalus (π = 0.0011). There were shared haplotypes among the three sampling sites for T. brevicephalus (Kka, Kal, Rur), but especially between samples from Lake Markakol’s tributary (Rur) and outflow (Kal).

Phylogenetic reconstruction of the relationships among the studied species of grayling estimated using the whole mtDNA CR and partial tRNA flanking region sequences. The represented topology was obtained in MrBayes and calculated using unique haplotypes only. The support values above and below nodes indicate Bayesian posterior probability (bpp) from MrBayes (%), and bootstrap support (bss) from RAxML, respectively. A star denotes 100 for both bpp and bss support values. Support values below 80% were omitted. Species are indicated on the right side of the tree and further highlighted using the same colors as shown in Fig. 1. Both unique and duplicate haplotypes for each of the tree focal species as well as T. svetovidovi are collapsed into triangles. The clades containing the three pairs of sister species discussed in this study are labeled A, B, & C
The time-calibrated phylogeny provided approximate divergence times among the focal taxa, all well within the Pleistocene epoch (Supplementary Material S3). The time to the most recent common ancestor (TMRCA) for T. brevirostris and T. brevicephalus was 0.5 MY (0.23–0.82), and for T. nikolskyi and T. svetovidovi 0.36 MY (0.13–0.62). The TMRCA for T. nikolskyi and the clade containing both T. brevirostris and T. brevicephalus was 1.1 MY (0.56–1.69), and for the one sister taxon pair inhabiting the same river drainage T. baicalensis and T. nigrescens the TMRCA was 0.18 MY (0.05–0.34).
The haplotype network indicated a close relationship between T. brevirostris and T. brevicephalus. Thymallus brevirostris revealed 18 haplotypes among 35 sampled individuals and T. brevicephalus 10 haplotypes among 32 sampled individuals (Fig. 4). The most frequent haplotype of T. brevicephalus was shared by individuals from the two sample sites directly connected to Lake Markakol (Rur and Kal), although both sample sites also had private (i.e. unique to the population sample) haplotypes. Five haplotypes were found exclusively in Kka, which is not directly connected to Lake Markakol, and one haplotype was shared between the Kka and the lake’s outflow (Kal). The genetic relationships among haplotypes for both T. nikolskyi and T. brevirostris and their corresponding geographic distribution did not support any clear phylogeographic structure in either species.

TCS 95% parsimony network(s) of all haplotypes from the three focal species, T. nikolskyi, T. brevicephalus, and T. brevirostris. Indels were coded as a 5th character state, small dots represent missing haplotypes, and circle size is proportional to haplotype frequencies
Population genetic analysis
Individual sample locations for T. brevirostris (Khg, Kht, Khv, Tol, Ach) revealed no statistically significant deviation from HWE proportions or LE. The mean expected heterozygosity was high across all sample locations and ranged from 0.67 for Lake Khoton (Kht) to 0.81 for the Khovd River (Khv). FIS values were distributed around zero except for the Lake Tolbo sample (Tol) (FIS = − 0.153), which indicates an excess of heterozygotes for this population sample (Table 4). Pairwise genetic differentiation (FST) for most pairwise site comparisons were low (0.010 to 0.028), and moderate (0.075) for the comparison of Kht and Tol (Table 5). Pairwise RST values for these same comparisons were generally higher but also highly variable compared to FST values (Table 5).
Similarly, for T. brevicephalus there was no deviation from HWE or LE for individual sample locations (Kka, Rur, Kal), and mean expected heterozygosity varied from 0.69 (Kal) to 0.83 (Kka) (Table 4). Kal and Rur displayed low genetic differentiation (FST = 0.037) while moderate levels of differentiation occurred between the two populations associated with Lake Markakol and Kka (FST = 0.129–0.165). The RST value (0.039) between the two Lake Markakol populations (Rur and Kal) was nearly identical to the FST value whereas RST values for all other pairwise comparisons were considerably higher, reaching 0.434 for the Kka vs. Kal comparison. All pairwise differentiation statistics (FST and RST) for T. brevicephalus were statistically significant (P < 0.05), supporting population substructure.
The mean FST between T. brevirostris samples from Western Mongolia and T. brevicephalus samples from the upper Irtysh River drainage was 0.25 whereby the individual values for each population varied widely (0.164–0.317). The mean FST between T. brevicephalus samples and the single sample of T. nikolskyi (Biy) was 0.190 (0.156–0.220).
The divergence among the putative species and different drainage systems is also seen in the FCA diagrams. All T. brevicephalus individuals cluster together, along the first axis (4.94% of variance) while individuals of T. nikolskyi and T. brevirostris formed their clusters along the second axis (4.37%) (Fig. 5a). The third axis of the FCA distinguishes the two population samples of T. brevicephalus associated with Lake Markakol (Rur and Kal) from individuals sampled from Kka (Fig. 5b). The differentiation between Lake Markakol (Rur and Kal) and Kka is also supported by the Bayesian clustering analysis (for K = 4). STRUCTURE runs indicated a large change in the posterior probabilities between K = 3 and K = 4 thus supporting K = 3 as the most optimal solution (following Evanno et al., 2005), with each species in a cluster (data not shown). However, there was also a significant peak at K = 4, with this additional group corresponding to a distinct population sample. Thus we display this four-cluster model (Fig. 6a), revealing that T. brevicephalus from Lake Markakol (Kal and Rur) and T. brevicephalus from Kara-Kaba River (Kka) form two separate clusters (dark and light red). Assignment probabilities (Q values) at K = 4 remained above 78% for all individuals, with little evidence of admixture between the four groups.

a First two axes of a Factorial Correspondence Analysis (FCA) based on the presence and absence of microsatellite alleles across seven loci for all three focal species. b second and third axes of this same analysis

a Graph of mean Q values from five independent runs of STRUCTURE using K = 4 and seven loci for all sample locations of the three focal species T. brevicephalus, T. brevirostris, and T. nikolskyi. b Graph of mean Q values from five independent runs of STRUCTURE using K = 2 using 10 loci for the three population samples (Kka, Kal, Rur) of T. brevicephalus
Extending our data set to 10 loci for the three T. brevicephalus populations only, produced the same clustering at K = 2 (Fig. 6b) as with the seven loci, with individuals of Kal and Rur in one cluster (bright red) and individuals of Kka in a second cluster (orange), with all individuals having a membership coefficient of > 90%.
Demographic modeling of Irtysh River drainage
The local logistic linear regression supported Scenario 1 (Fig. 2), as the best fit for the data, with an initial split (t2) of Kka from an ancestral population, followed by a change in effective population sizes (t12) along the branch leading eventually to a split between Rur and Kal (t1) (Fig. 2). The first split (t2) in this scenario was estimated to occur 2 × 104 generations ago, and approximately 1 × 104 generations ago (t12), the remaining ancestral population underwent a demographic change leading to the final split (t1) of the two sample populations (Rur and Kal) 5 × 103 generations ago. These results are concordant with the differentiation between the Lake Markakol (Rur and Kal) and Kara-Kaba River (Kka) populations. The effective population size was estimated at 2 × 104 for all populations. An overview of all priors and the posterior probabilities of all parameters for Scenario 1 is provided in Supplementary Materials S4.
Discussion
Phylogenetic and phylogeographic overview
The mtDNA-based phylogenetic results conform to existing taxonomic descriptions (Dyldin et al., 2017). Haplotypes from each of the three putative focal species of this study (T. brevicephalus, T. brevirostris and T. nikolskyi,) form reciprocally monophyletic clades with their respective sister taxon. The distributions of these three taxa correspond to three rather small adjacent freshwater ecoregions: upper Irtysh (Ecoregion ID: 603), Western Mongolia (622), and Chuya (604), following Abell et al. (2008). Nonetheless, the phylogenetic split pattern does not follow the contemporary river drainage networks. The distributions of the focal species, along with T. svetovidovi, the sister taxon to T. nikolskyi, appear to have been strongly influenced by paleohydrological events. Here we would like to point out that there is an inherent weakness in drawing inferences based on phylogenetic analysis of mtDNA due to its maternal inheritance (e.g. see Ballard & Whitlock, 2004). However, our multi-locus population genetic analyses are highly supportive of our phylogenetic results, as there is little to no sign of introgression or hybridization among the three focal species. This, combined with the fact that these species are all found in allopatry with no contemporary hydrological connection between their distribution areas, results in a relatively high likelihood that the mtDNA phylogeny reflects the population history of these species. We briefly review knowledge of these species in light of our new data as well as the existing paleohydrological information, in an attempt to assess the role that these processes may have played in shaping their current distributions.
Thymallus nikolskyi: Upper Ob grayling
Thymallus nikolskyi is the sister species of T. svetovidovi of the upper Enisei River drainage in northwestern Mongolia. The Upper Ob grayling is not widely recognized as a distinct species outside of the Russian literature and has been often synonymized with Arctic grayling (T. arcticus) or recognized as a subspecies of Arctic grayling. However, as shown here, the lineage is 3.6% divergent from T. arcticus (Fig. 3; Table 3) and is phenotypically distinct (Kaschenko, 1899; Romanov et al., 2016). Severin & Zinoviev (1982) reported on a minor karyotypic difference between the grayling populations of the lower reaches of the Ob (98–100 chromosomes), presumably T. arcticus, and those of the upper reaches (100–102), presumably T. nikolskyi. Additionally, nearly three-quarters of the 45 morphological characters investigated revealed statistically significant differences between populations from these two reaches of the Ob River drainage (Severin & Zinoviev, 1982).
Taxonomic revisions of Svetovidov (1936) and Berg (1948) reported that only T. arcticus inhabit the Ob River drainage, with the holotype of the species stemming from an Ob River tributary, the Sob River, which joins the Ob ca. 70 km before it drains into the Kara Sea. This locality is over 3000 km river distance from the headwaters of the Ob (rivers Katun, Biya and Tom) comprising the type localities of T. nikolskyi. This pattern of T. arcticus inhabiting the lower course of a major Palearctic river system, and the headwaters harboring other grayling species, is consistent across the Ob, Enisei and Lena river drainages. Thus, T. arcticus in the Lena River is replaced in the headwaters by T. baicalolenensis (Knizhin et al., 2006a, Weiss et al., 2006); and T. arcticus in the lowermost Enisei River is replaced by T. baicalensis in most of the Enisei and Lake Baikal catchments (Knizhin et al., 2006b; Weiss et al., 2007). Of these three basins and their corresponding species pairs, only the two taxa in the Lena River catchment (T. arcticus and T. baicalolenensis) show a sister taxon relationship, possibly reflecting within-basin cladogenesis.
Thymallus brevicephalus: Markakol grayling
Like T. nikolskyi, T. brevicephalus receives little attention outside of the Russian literature. Based on our mtDNA data, its divergence from T. brevirostris occurring in the endorheic basin of Western Mongolia corresponds to about 500,000 years, whereby its divergence to T. nikolskyi sampled from the upper Ob River drainage corresponds to approximately 1.1 MY. The moderately high FST values for the seven microsatellite between T. nikolskyi and T. brevicephalus (0.156–0.220) likely underestimate divergence due to homoplasy, which is a common occurrence for microsatellites from highly divergent lineages (see Queney et al., 2001), especially if the populations are large, as is likely the case here. Accordingly, the RST values, which account for allele size differences (see Slatkin, 1995 and discussion in Sefc et al., 2007), are comparatively higher, ranging from 0.245 to 0.312.
Thymallus brevicephalus was described based on a considerably different head morphology compared to either T. brevirostris or other grayling found in the Ob River drainage (Mitrofanov, 1971; Mitrofanov et al., 1986). Little else has been published on this species but it is clear that, opposed to T. brevirostris, it lacks dentition or large jaws and is a rather small-sized grayling (mostly less than 30 cm) that primarily feeds on benthic invertebrates (e.g. Gammarus and Trichoptera) and, seasonally, terrestrial insects (Coleoptera and Formicidae) (Mitrofanov et al., 1986). Thymallus brevicephalus has a restricted distribution, centered on Lake Markakol, although the exact extent of its distribution is not known with certainty. Lake Markakol (Rur, Kal) and Kara-Kaba river (Kka) populations of T. brevicephalus are well-differentiated, with high RST-and FST-values, with the most likely ABC scenario supporting a split at approximately 100,000 years (generation time of 5 years) with no likely subsequent contact. The split between the inflow and outflow sampling sites (Rur and Kal) equates to approximately 25,000 years. While we assume that all three of these population samples belong to one species, we lack sufficient phenotypic data to definitively determine if the Kka sample can also be phenotypically assigned to T. brevicephalus.
Thymallus brevirostris: Mongolian grayling
In contrast to the other two focal species of this study, T. brevirostris has received considerable attention reaching back over a century (see Knizhin et al., 2008 and citations therein). In addition to its well-documented distribution in Western Mongolia and the Tuva Republic, Russia, several reports exist that the species also occurs south of the Altai Mountain range in the upper Irtysh River drainage of northern China. These reports began with the description of Phylogephyra altaica Boulenger, 1898 south of the Altai, which, based on morphological features, has been synonymized with T. brevirostris (Knizhin, 2009). The present-day occurrence of T. brevirostris in this region is poorly documented. Thymallus brevirostris is primarily known as the only member of the genus bearing relatively well-developed teeth on both the upper and lower jaws, vomer, palatine and tongue (Knizhin, 2009). The species is generally described as a large-growing piscivorous inhabitant of the species’ poor lakes of Western Mongolia. However, surveys of the species’ range have consistently reported a smaller-sized, insectivorous cogener T. cf. arcticus, sharing habitat or living close to Mongolian grayling (Knizhin et al., 2008 and citations therein). These fish, however, at least from Western Mongolia, and based on mtDNA control region sequences, belong to the same monophyletic clade as T. brevirostris (Knizhin et al., 2008), with no apparent genetic structure. Our current data set further supports these results with the application of multi-locus population genetic data (e.g. Fig. 5). Without such population genetic data, one could not have excluded that the smaller-sized, insectivorous grayling in the region form a distinct genetic unit, or that phylogenetic relationship between small insectivorous and large-piscivorous grayling based on mtDNA is merely the result of one or more hybridization events. Nonetheless, the high phenotypic variability of grayling inhabiting Western Mongolia still requires further explanation, and the genetic resolution of the data presented here (mtDNA control region + 7 microsatellite loci) is limited and may not be able to detect most recent genetic divergence, or other more complex genetic-environmental phenomena resulting in one or more phenotypic variants.
Thymallus nigrescens: Chuvsgol grayling and T. baicalensis: Enisei grayling
In contrast to the geographic and phylogenetic relation of the three focal species, T. nigrescens and T. baicalensis show a close sister relationship (Fig. 3) and occur in the same major river drainage (Enisei). It has been suggested that T. nigrescens is an ecotype of T. baicalensis (Kaus et al., 2019). However, the current distribution of T. baicalensis has been established to be the result of a cataclysmic paleohydrological event. As described in Koskinen et al. (2002), Lake Baikal was colonized from Enisei River drainage after the Angara River breakout between 110,000 and 130,000 years ago. This event began with the tectonically caused isolation of the lake, subsequent rising water levels, and an earthquake and/or landslide resulting in a massive flood that established Lake Baikal’s contemporary outlet to the Enisei River drainage, via the Angara River (Arzhannikov et al., 2018; Fig. 1). Enisei grayling expanded up into the Selenga River of Mongolia including Lake Chuvsgol in Northwestern Mongolia. While support for a single expansion event into Lake Chuvsgol was supported by both microsatellites and mtDNA (Koskinen et al., 2002) the results of a recent Next-Generation-Sequencing (NGS) study has led to hypothesize a more complex colonization of Lake Chuvsgol (Roman et al., 2018). Additionally, evidence is provided for resource partitioning within T. nigrescens in Lake Chuvsgol, which may indicate incipient speciation (Olson et al., 2019).
Biogeography and paleohydrology
While the full geographic ranges of the six Thymallus species occurring in the Altai-Sayan mountain region (Fig. 1 and clades A–C in Fig. 3) are not known with certainty, it appears that all have been fundamentally affected by major paleohydrological events. From the mega-flood events pictured in Fig. 1, only the Angara breakout (e in Fig. 1) has a timing relevant to the contemporary distribution of two taxa, T. baicalensis and T. nigrescens. The sister relationship of T. svetovidovi and T. nikolskyi must stem from a historical connection between the uppermost Ob River drainage and the headwaters of the Enisei River in northern Mongolia (or the Tuva Republic). Both the “Kas-Ket” (a in Fig. 1) and “Sayan” (d in Fig. 1) mega-flood events (Komatsu et al., 2016) propose connections between the Enisei and Ob river drainages. Furthermore, there is evidence that T. baicalensis, whose distribution was thought to be limited to the Enisei River drainage, occurs in at least one right tributary of the Ob River drainage, as revealed by recent analysis based on mitogenomes across the entire genus (Weiss et al., unpublished data). This occurrence may be indeed directly linked to the “Kas-Ket” mega-flood event (a in Fig. 1).
These events dating back to the late Pleistocene or early Holocene are too young to explain the common ancestry of taxa that are approximately 360,000 years divergent. Thus, we take these events as simply exemplary for the dynamics of the region, noting that the traces of yet older floods along the same corridors as those shown here would have likely been largely erased by more recent ones. Additional biogeographic support for these connections can be seen with the co-occurrence of both Oreoleuciscus potanini (Kessler, 1879) and O. humilis (Warpachowski, 1899) in the drainages of the upper Ob River and the endorheic basin of Western Mongolia (Golubtsov et al., 1999; Bogutskaya, 2001; Slynko & Dgebuadze, 2009; Slynko & Borovikova, 2012).
While the “Altai” mega-flood (see b in Fig. 1) (Komatsu et al., 2016), as well as the evidence of repeated outbursts from the paleolake in the Khindiktik-Kol basin through the Mogen-Buren valley (see c in Fig. 1) (Agatova et al., 2015; Agatova & Nepop, 2019), hypothesized paleohydrological corridors between the endorheic basin of Western Mongolia and the upper Ob River drainage, it is not clear how these events could have influenced the contemporary distribution of Thymallus in the region.
We lack a hypothesis for the origin of T. brevicephalus as it is the sister clade to T. brevirostris and not closely related to other Thymallus taxa found in the Ob River drainage (T. arcticus, T. nikolskyi and T. baicalensis). The most likely connection between the now isolated basins of Western Mongolia and the upper Irtysh drainage would be a corridor across the Altai Mountains via the headwaters of the uppermost Irtysh River (referred to as the Black Irtysh). This region has also undergone substantial tectonic activity as the Black Irtysh is thought to have been isolated from the lower Irtysh River (or White Irtysh) with the ancient Lake Zaysan being the border. Thus, rivers on the south slopes of the Altai Mountains are hypothesized to have drained south into the Lake Manas catchment in China before being diverted along fault lines to the west into the contemporary upper Irtysh River corridor (Yao & Li, 2010; Jolivet et al., 2013). Here again, the co-occurrence of another genus, Triplophysa lends support to such a transgression. The genus is primarily found on the Tibetan Plateau (Yang et al., 2019) but also in the upper Irtysh drainage (Yang et al., 2016) as well as in the Uvs Nuur and Khovd River drainages of Western Mongolia (Kottelat, 2006; Prokofiev, 2006). Thus, an historical hydrological colonization route through the Altai Mountains between the endorheic basin of Western Mongolia and the upper Irtysh River drainage of northern China and Kazachstan seems plausible.
Several GenBank entries for Thymallus from the Irtysh River drainage in China are inconclusive, as two entries of mtDNA COI sequences labeled Thymallus arcticus arcticus sampled from an unknown location in the upper Irtysh River (GenBank nos. KT716357 and KT716358, Yang et al., 2016) group with our data from T. brevicephalus from Lake Markakol, and a mitogenome of T. brevirostris (GenBank no. KJ866486, Ma et al., 2016) lists the Altai City market as the source of the sample, leaving some doubt as to whether the fish came from local waters or was transported for sale from nearby Mongolia. Thus, questions remain on whether T. brevirostris currently occurs south of the Altai Mountains in China. Equally intriguing is the question of whether the genetic lineage corresponding to T. brevicephalus in our data can be found in the uppermost headwaters of the Black Irtysh, a part of which is found in Western Mongolia.
Future outlook
While this contribution adds considerable knowledge on grayling diversity and relationships in the Altai-Sayan mountain region, several issues need further clarification. For T. brevirostris, further sampling south of the Altai Mountains in China will be needed to determine if the species occurs there. Deeper genome-level sequencing will be needed to elucidate the genetic mechanism responsible for the phenotypic variability exhibited by the taxon. For the upper Ob (inclusive of the large Irtysh sub-drainage and specifically Lake Markakol) better phenotypic data is needed to clarify both taxon delineation and the full geographic distributions of T. nikolskyi and T. brevicephalus. Here we note that T. brevicephalus has undergone serious declines in Lake Markakol, due to overfishing (pers comm. M. Baimukanov), and both its phenotypic distinctiveness as well as its spawning behavior (reportedly entering tributaries and outflows in early spring) should be better investigated and used to support a conservation management plan for the species, especially considering that the entire lake system is a nature reserve. It is also likely that T. baicalensis and T. nikolskyi come into contact in one or more right tributaries of the Ob, and if one of these zones could be located one can test for reproductive isolation, such as has been clearly shown for other Asian grayling species in contact zones of the lower Enisei River drainage (Weiss et al., 2007) and the Bureya River of the Amur River (Weiss et al., 2020). In those studies, sympatric species were clearly of allopatric origin, similar to what we describe here for the Altai-Sayan mountain region. Paleohydrological processes that both isolate and reconnect lineages have played a major role in cladogenesis and distribution of grayling species throughout Eurasia.
Lastly, grayling inhabiting the upper Enisei and its tributaries in Russia, just north of Mongolia, have been poorly investigated and their taxonomic status remains unclear. Two taxa, listed as subspecies of T. arcticus, have been described in that region based on morphological data only (Grundriser, 1967, 1979). Considering the complexity and lineage diversity that we have shown to occur in the Altai-Sayan mountain region these taxa require reinvestigation, considering both their phenotype and their phylogenetic relationships.
Change history
29 June 2020
Due to an error��during the production process, the incorrect handling editor was displayed in the original publication. The handling editor mentioned in the original paper works at the same department as some of the authors, and he concluded that this might constitute a conflict of interest. The paper was therefore handled by one of the associate editors in chief. The correct footnote is published here and should be treated as definitive:
References
Abell, R., M. L. Thieme, C. Revenga, M. Bryer, M. Kottelat, N. Bogutskaya, B. Coad, N. Mandrak, S. C. Balderas, W. Bussing, M. L. J. Stiassny, P. Skelton, G. R. Allen, P. Unmack, A. Naseka, R. Ng, N. Sindorf, J. Robertson, E. Armijo, J. V. Higgins, T. J. Heibel, E. Wikramanayake, D. Olson, H. L. López, R. E. Reis, J. G. Lundberg, M. H. Sabaj Pérez & P. Petry, 2008. Freshwater ecoregions of the world: a new map of biogeographic units for freshwater biodiversity conservation. BioScience Narnia 58: 403–414.
Agatova, A. R. & R. K. Nepop, 2019. Pleistocene fluvial catastrophes in now arid NW areas of Mongolian Inland drainage basin. Global and Planetary Change 175: 211–225.
Agatova, A. R., R. K. Nepop & M. Brannikova, 2015. Outburst floods of ice-dammed lakes of the SW Tuva, in Siberia. Zeitschrift für Geomorphologie 59: 159–175.
Antonov, A. L., 2004. A new species of grayling Thymallus burejensis sp. nov. (Thymallidae) from the Amur Basin. Journal of Ichthyology 44: 401–411.
Arzhannikov, S. G., A. V. Ivanov, A. V. Arzhannikova, E. I. Demonterova, J. D. Jansen, F. Preusser, V. S. Kamenetsky & M. B. Kamenetsky, 2018. Catastrophic events in the quaternary outflow history of Lake Baikal. Earth-Science Reviews 177: 76–113.
Avise, J. C., 2007. Twenty-Five Key Evolutionary Insights from the Phylogeographic Revolution in Population Genetics. Phylogeography of Southern European Refugia. Springer, Dordrecht: 7–21.
Ballard, J. W. O. & M. C. Whitlock, 2004. The incomplete natural history of mitochondria. Molecular Ecology 13: 729–744.
Belkhir, K., 1996–2004. Genetix 4.05, logiciel sous Windows TM pour la genetique des populations. Laboratoire Genome, Populations, Interactions, CNRS UMR 5000, Universite de Montpellier II. http://www.genetix.univ-montp2.fr/genetix/genetix.htm.
Berg, L. S., 1948. Freshwater Fishes of the USSR and Adjacent Countries, Part 1, 4th ed. Akad. Nauk SSSR, Moscow (in Russian).
Bernatchez, L., R. Guyomard & F. Bonhomme, 1992. DNA sequence variation of the mitochondrial control region among geographically and morphologically remote European brown trout Salmo trutta populations. Molecular Ecology 1: 161–173.
Bogutskaya, N. G., 2001. A revision of altai osmans of the genus Oreoleuciscus (Leuciscinae, Cyprinidae) with a Description of a new species, O. angusticephalus, from River Kobdo (Hovd) system, West Mongolia. Proceedings of the Zoological Institute St. Petersburg 287: 5–43.
Bohorquez, P., P. A. Carling & J. Herget, 2015. International Geology Review Dynamic simulation of catastrophic late Pleistocene glacial-lake drainage, Altai Mountains, central Asia Dynamic simulation of catastrophic late Pleistocene glacial-lake drainage, Altai Mountains, central Asia. International Geology Review 58: 1795–1817.
Buj, I., Z. Marčić, M. Ćaleta, R. Šanda, M. F. Geiger, J. Freyhof, A. Machordom & J. Vukić, 2017. Ancient connections among the European rivers and watersheds revealed from the evolutionary history of the genus Telestes (Actinopterygii; Cypriniformes). PLoS ONE 12: e0187366.
Carvajal-Quintero, J., F. Villalobos, T. Oberdorff, G. Grenouillet, S. Brosse, B. Hugueny, C. Jézéquel & P. A. Tedesco, 2019. Drainage network position and historical connectivity explain global patterns in freshwater fishes’ range size. Proceedings of the National Academy of Sciences of the United States of America 116: 13434–13439.
Clement, M., D. Posada & K. A. Crandall, 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659.
Cornuet, J.-M., P. Pudlo, J. Veyssier, A. Dehne-Garcia, M. Gautier, R. Leblois, J.-M. Marin & A. Estoup, 2014. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism. DNA sequence and microsatellite data. Bioinformatics 30: 1187–1189.
Craw, D., C. Burridge, L. Anderson & J. M. Waters, 2007. Late Quaternary river drainage and fish evolution, Southland, New Zealand. Geomorphology 84: 98–110.
Csilléry, K., M. G. B. Blum, O. E. Gaggiotti & O. François, 2010. Approximate Bayesian computation (ABC) in practice. Trends in Ecology & Evolution 25: 410–418.
Darriba, D., G. L. Taboada, R. Doallo & D. Posada, 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9: 772.
de Bruyn, M., L. Rüber, S. Nylinder, B. Stelbrink, N. R. Lovejoy, S. Lavoué, H. H. Tan, E. Nugroho, D. Wowor, P. K. L. Ng, M. N. Siti Azizah, T. Von Rintelen, R. Hall & G. R. Carvalho, 2013. Paleo-drainage basin connectivity predicts evolutionary relationships across three southeast asian biodiversity hotspots. Systematic Biology 62: 398–410.
Dias, M. S., T. Oberdorff, B. Hugueny, F. Leprieur, C. Jézéquel, J.-F. Cornu, S. Brosse, G. Grenouillet & P. A. Tedesco, 2014. Global imprint of historical connectivity on freshwater fish biodiversity. Ecology Letters 17: 1130–1140.
Diggs, M. D. & W. R. Ardren, 2008. Characterization of 12 highly variable tetranucleotide microsatellite loci for Arctic grayling (Thymallus arcticus) and cross amplification in other Thymallus species. Molecular Ecology Resources 8: 828–830.
Drummond, A. J. & A. Rambaut, 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7: 214.
Drummond, A. J., S. Y. W. Ho, M. J. Phillips & A. Rambaut, 2006. Relaxed phylogenetics and dating with confidence. PLoS Biology 4: e88.
Durand, J. D., H. Persat & Y. Bouvet, 1999. Phylogeography and postglacial dispersion of the chub (Leuciscus cephalus) in Europe. Molecular Ecology 8: 989–997.
Dyldin, Y., V. Hanel, V. Romanov & J. Plesnik, 2017. A review of the fish genus Thymallus (Pisces: Salmoniformes, Salmonidae, Thymallinae) with taxonomic notes. Bulletin Lampetra 8: 103–126.
Earl, D. A. & B. M. vonHoldt, 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetic Resources 4: 359–361.
Estoup, A., P. Jarne & J.-M. Cornuet, 2002. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular Ecology 11: 1591–1604.
Evanno, G., S. Regnaut & J. Goudet, 2005. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology 14: 2611–2620.
Excoffier, L. & H. E. L. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology 10: 564–567.
Frick, R., W.N. Eschmeyer, & R. van der Laan, 2020. Eschmeyer’s catalog of fishes: general, species, references. http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp. Accessed Mar 2, 2020.
Froufe, E., S. Alekseyev, I. Knizhin, P. Alexandrino & S. Weiss, 2003. Comparative phylogeography of salmonid fishes (Salmonidae) reveals late to post-Pleistocene exchange between three now-disjunct river basins in Siberia. Diversity and Distributions 9: 269–282.
Froufe, E., I. Knizhin & S. Weiss, 2005. Phylogenetic analysis of the genus Thymallus (grayling) based on mtDNA control region and ATPase 6 genes, with inferences on control region constraints and broad-scale Eurasian phylogeography. Molecular Phylogenetics and Evolution 34: 106–117.
Froufe, E., S. Alekseyev, P. Alexandrino & S. Weiss, 2008. The evolutionary history of sharp- and blunt-snouted lenok (Brachymystax lenok (Pallas, 1773)) and its implications for the paleo-hydrological history of Siberia. BMC Evolutionary Biology 8: 40.
Gernhard, T., 2008. The conditioned reconstructed process. Journal of Theoretical Biology 253: 769–778.
Gillespie, A. R., R. M. Burke, G. Komatsu & A. Bayasgalan, 2008. Late Pleistocene glaciers in Darhad Basin, northern Mongolia. Quaternary Research 69: 169ΓÇô187.
Golubtsov, A. S., P. B. Berendzen & C. A. Annett, 1999. Morphological variation and taxonomic status of Altai osmans, Oreoleuciscus, from the upper reaches of the Ob River system. Journal of Fish Biology 54: 878–899.
Goudet, J., 2001. FSTAT, a program to estimate and test gene diversities and fixation indices, version 2.9.3. http://www2.unil.ch/popgen/softwares/fstat.htm.
Griffiths, D., 2015. Connectivity and vagility determine spatial richness gradients and diversification of freshwater fish in North American and Europe. Biological Journal of the Linnean Society 116: 773–786.
Grosswald, M. G., 1998. New approach to the ice age paleohydrology of northern Eurasia. In Benito, G., V. R. Baker & K. J. Gregory (eds), Paleohydrology and Environmental Change. Wiley, Chichester: 199–214.
Gundriser, A. N., 1967. About a new subspecies of Siberian grayling from the basin of the big Yenisei [= O novom podvide sibirskogo khariusa iz basseyna Bol’shego Yeniseya]. Uchenyye zapiski Tomskogo Ordena Trudovogo Krasnogo Znameni Gosudarstvennogo Universiteta im. V.V. Kuybysheva 53: 79–94 (in Russian).
Gundriser, A. N., 1979. Dentated Siberian grayling – Thymallus arcticus dentatus subsp. Nova [= Zubastyy sibirskiy kharius – Thymallus arcticus dentatus Subsp. Nova]. In New data on the fauna and flora of Siberia [= Novye dannye o faune i flore Sibiri]. Tomsk: Publishing house of Tomsk University: 15–22 (in Russian).
Hasegawa, M., H. Kishino & T. Yano, 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22: 160–174.
Ivanov, A. V., E. I. Demonterova, L. Z. Reznitskii, I. G. Barash, S. G. Arzhannikov, A. V. Arzhannikova, C.-H. Hung, S.-L. Chung & Y. Iizuka, 2016. Catastrophic outburst and tsunami flooding of Lake Baikal: U-Pb detrital zircon provenance study of the Palaeo-Manzurka megaflood sediments. International Geology Review 58: 1818–1830.
Jolivet, M., G. Heilbronn, C. Robin, L. Barrier, S. Bourquin, Zh Guo, Y. Jia, L. Guerit, W. Yang & B. Fu, 2013. Reconstructing the Late Palaeozoic—Mesozoic topographic evolution of the Chinese Tian Shan: available data and remaining uncertainties. Advances in Geosciences 37: 7–18.
Junge, C., C. R. Primmer, L. A. Vøllestad & E. H. Leder, 2010. Isolation and characterization of 19 new microsatellites for European grayling, Thymallus thymallus (Linnaeus, 1758), and their cross-amplification in four other salmonid species. Conservation Genetics Resources 2: 219–223.
Kaschenko, N. F., 1899. Results of Zoological Expedition to Altai in 1898 Vertebrates. Tomsk University, Tomsk: 131–155 (in Russian).
Katoh, K. & D. M. Standley, 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780.
Kaus, A., S. Michalski, B. Hänfling, D. Karthe, D. Borchardt & W. Durka, 2019. Fish conservation in the land of steppe and sky: evolutionarily significant units of threatened salmonid species in Mongolia mirror major river basins. Ecology and Evolution 9: 3416–3433.
Knizhin, I. B., 2009. Graylings (Thymallus Cuvier, 1829) of the Holarctic (Systematics, Phylogeography, and Ecological features). Doctoral thesis, Severtsov Institute of Ecology and Evolution, Russian Academy of Science, Moscow: 268 (in Russian).
Knizhin, I. B. & S. J. Weiss, 2009. A new species of grayling Thymallus svetovidovi sp. nova (Thymallidae) from the Yenisei basin and its position in the genus Thymallus. Journal of Ichthyology 49: 1–9.
Knizhin, I. B., A. F. Kirillov & S. J. Weiss, 2006a. On the diversity and taxonomic status of graylings (Thymallus, Thymallidae) from the Lena River. Journal of Ichthyology 46: 234–246.
Knizhin, I. B., S. J. Weiss & S. Sušnik, 2006b. Graylings of Baikal Lake basin (Thymallus, Thymallidae): diversity of forms and their taxonomic status. Journal of Ichthyology 46: 418–435.
Knizhin, I. B., S. J. Weiss, B. E. Bogdanov, T. Kopun & O. V. Muzalevskaya, 2008. Graylings (Thymallidae) of water bodies in western Mongolia: morphological and genetic diversity. Journal of Ichthyology 48: 714–735.
Komatsu, G., V. R. Baker, S. G. Arzhannikov, R. Gallagher, A. V. Arzhannikova, A. Murana & T. Oguchi, 2016. Catastrophic flooding, palaeolakes, and late Quaternary drainage reorganization in northern Eurasia. International Geology Review 58: 1693–1722.
Koskinen, M. T., I. Knizhin, C. R. Primmer, C. Schlötterer & S. Weiss, 2002. Mitochondrial and nuclear DNA phylogeography of Thymallus spp. (grayling) provides evidence of ice-age mediated environmental perturbations in the world’s oldest body of fresh water, Lake Baikal. Molecular Ecology 11: 2599–2611.
Kottelat, M., 2006. Fishes of Mongolia A Check-List of the Fishes Known to Occur in Mongolia with Comments on Systematics and Nomenclature. Environmental and Social Development Sector, East Asia and Pacific Region. The World Bank, Washington: 104.
Kumar, S., G. Stecher, M. Li, C. Knyaz & K. Tamura, 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution 35: 1547–1549.
Lerceteau-Köhler, E., U. Schliewen, T. Kopun & S. Weiss, 2013. Genetic variation in brown trout Salmo trutta across the Danube, Rhine, and Elbe headwaters: a failure of the phylogeographic paradigm? BMC Evolutionary Biology 13: 176.
Levin, B. A., A. A. Gandlin, E. S. Simonov, M. A. Levina, A. E. Barmintseva, B. Japoshvili, N. S. Mugue, L. Mumladze, N. J. Mustafayev, A. N. Pashkov, H. R. Roubenyan, M. I. Shapovalov & I. Doadrio, 2019. Phylogeny, phylogeography and hybridization of Caucasian barbels of the genus Barbus (Actinopterygii, Cyprinidae). Molecular Phylogenetics and Evolution 135: 31–44.
Librado, P. & J. Rozas, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Ma, B., H. Jiang, P. Sun, J. Chen, L. Li, X. Zhang & L. Yuan, 2016. Phylogeny and dating of divergences within the genus Thymallus (Salmonidae: Thymallinae) using complete mitochondrial genomes. Mitochondrial DNA 27: 3602–3611.
Margold, M., J. D. Jansen, A. T. Codilean, F. Preusser, A. L. Gurinov, T. Fujioka & D. Fink, 2018. Repeated megafloods from glacial Lake Vitim, Siberia, to the Arctic Ocean over the past 60,000 years. Quaternary Science Reviews 187: 41–61.
Matamoros, W., C. W. Hoagstrom, J. F. Schaefer & B. R. Kreiser, 2016. Fish fauna provinces of the conterminous United States of America reflect historical geography and familial composition. Biological Reviews 91: 813–832.
Miller, M. A., W. Pfeiffer, & T. Schwartz, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In 2010 Gateway Computing Environments Workshop (GCE). IEEE: 1–8.
Mitrofanov, V. P., 1971. Grayling of the Lake Markakul. Biological Sciences 1: 104–112 (in Russian).
Mitrofanov, V. P., G. M. Dukravets, N. E. Peseridi, & A. N. Poltorykhina, 1986. Fishes of the Kazakhstan. V. 1. Izdatelstvo GYLYM, Alma-Ata: Nauka, 271 pp (in Russian).
Near, T. J. & B. P. Keck, 2005. Dispersal, vicariance, and timing of diversification in Nothonotus darters. Molecular Ecology 14: 3485–3496.
Nei, M. & W. H. Li, 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences of the United States of America 76: 5269–5273.
Nesbo, C. L., T. Fossheim, L. A. Vollestad & K. S. Jakobsen, 1999. Genetic divergence and phylogeographic relationships among European perch (Perca fluviatilis) populations reflect glacial refugia and postglacial colonization. Molecular Ecology 8: 1387–1404.
Olson, K. W., T. J. Krabbenhoft, T. R. Hrabik, B. Mendsaikhan & O. P. Jensen, 2019. Pelagic–littoral resource polymorphism in Hovsgol grayling Thymallus nigrescens from Lake Hovsgol, Mongolia. Ecology of Freshwater Fish 28: 411–423.
Pritchard, J. K., M. Stephens, & P. Donnelly, 2000. Inference of population structure using multilocus genotype data. http://www.stats.ox.ac.uk/pritch/home.html.
Prokofiev, A. M., 2006. Two new species of the loach genus Triplophysa Rendahl, 1933 from western Mongolia and northwestern China, with a key of the species from the interior drainages of Tien-Shan, Karakurum and Altai Mountains (Osteichthyes, Balitoridae, Nemacheilinae). Senckenbergiana Biologica 86: 235–259.
Queney, G., N. Ferrand, S. Weiss, F. Mougel & M. Monnerot, 2001. Stationary distributions of microsatellite loci between divergent population groups of the European rabbit (Oryctolagus cuniculus). Molecular Biology and Evolution 18: 2169–2178.
Rambaut, A., 2012. FigTree v1.4. Molecular Evolution, Phylogenetics and Epidemiology. Edinburgh, UK. http://tree.bio.ed.ac.uk/software/figtree.
Rambaut, A., A. J. Drummond, D. Xie, G. Baele & M. A. Suchard, 2018. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Systematic Biology 67: 901–904.
Roman, I., Y. Bourgeois, J. Reyes-Velasco, O. P. Jensen, J. Waldman, S. Boissinot, N. York, S. I. Campus, A. Dhabi & U. A. Emirates, 2018. Contrasted patterns of divergence and gene flow among five fish species in a Mongolian rift lake following glaciation. Biological Journal of the Linnean Society 125: 1–11.
Romanov, V., Y. Lavrentiev & Y. Dyldin, 2016. On the morphology of the Siberian grayling (Thymallus arcticus (Pallas)) dwelling in the Kazim River Basin (Lower Ob River). Surgut Bulletin 3: 37–42 (in Russian).
Ronquist, F. & J. P. Huelsenbeck, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Sambrook, J., E. Fritsch & T. Maniatis, 1989. Molecular Cloning: A Laboratory Manual. Cold Harbor Spring Press, New York.
Schönhuth, S., M. J. Blum, L. Lozano-Vilano, D. A. Neely, A. Varela-Romero, H. Espinosa, A. Perdices & R. L. Mayden, 2011. Inter-basin exchange and repeated headwater capture across the Sierra Madre Occidental inferred from the phylogeography of Mexican stonerollers. Journal of Biogeography 38: 1406–1421.
Sefc, K. M., R. B. Payne & M. D. Sorenson, 2007. Genetic differentiation after founder events: an evaluation of FST estimators with empirical and simulationed data. Evolutionary Ecology Research 9: 21–39.
Severin, S. O. & E. A. Zinoviev, 1982. Karyotypes of isolated populations of Thymallus arcticus (Pallas) in the Ob river basin. Problems of Ichthyology 22: 27–35 (in Russian).
Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Slynko, Y. V. & E. A. Borovikova, 2012. Phylogeography of Altai osmans fishes (Oreoleuciscus sp., Cyprinidae, Pisces) inferred from nucleotide variation of the mitochondrial DNA cytochrome b gene. Russian Journal of Genetics 48: 618–627.
Slynko, Y. V. & Y. Y. Dgebuadze, 2009. Population-genetical analysis of Altai osmans (Oreoleuciscus, Cyprinidae) from water bodies of Mongolia. Journal of Ichthyology 49: 614–626.
Slynko, Y. V., B. Mendsaykhan & A. N. Kas’anov, 2010. On intraspecies forms of the Mongolian grayling (Thymallus brevirostris Kessl.) from Hoton Nur Lake (Western Mongolia). Journal of Ichthyology 50: 28–37.
Smith, G. R., 1992. Introgression in fishes: significance for paleontology, cladistics, and evolutionary rates. Systematic Biology 41: 41–57.
Snoj, A., S. Susnik, J. Pohar & P. Dovc, 1999. The first microsatellite marker (BFRO 004) for grayling, informative for its Adriatic population. Animal Genetics 30: 74–75.
Stamatakis, A., 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313.
Stepien, C. A., A. K. Dillon & M. D. Chandler, 1998. Genetic identity, phylogeography, and systematics of Ruffe Gymnocephalus in the North American Great Lakes and Eurasia. Journal of Great Lakes Research 24: 361–378.
Suchard, M. A. & A. Rambaut, 2009. Many-core algorithms for statistical phylogenetics. Bioinformatics Narnia 25: 1370–1376.
Sušnik, S., A. Snoj, D. Jesenšek & P. Dovč, 2000. Rapid communication: microsatellite DNA markers (BFRO010 and BFRO011) for grayling. Journal of Animal Science 78: 488–489.
Svenning, J. C., W. L. Eiserhardt, S. Normand, A. Ordonez & B. Sandel, 2015. The influence of Paleoclimate on present-day patterns in biodiversity and ecosystems. Annual Review of Ecology, Evolution, and Systematics 46: 551–572.
Svetovidov, A. N., 1936. European-Asiatic graylings (Genus Thymallus Cuvier), Study of the Institute of Zoology. Akademii Nauk SSSR 3: 183–301 (in Russian).
Templeton, A. R., K. A. Crandall & C. F. Sing, 1992. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132: 619–633.
Uiblein, F., A. Jagsch, W. Honsig-Erlenburg & S. Weiss, 2001. Status, habitat use, and vulnerability of the European grayling in Austrian waters. Journal of Fish Biology 59: 223–247.
Villeseon, P., 2007. FaBox: an online toolbox for FASTA sequences. Molecular Ecology Notes 7: 965–968.
Waters, J. M., C. P. Burridge & D. Craw, 2019. The lasting biological signature of Pliocene tectonics: reviewing the re-routing of Australia’s largest river drainage system. Journal of Biogeography 46: 1494–1503.
Weiss, S., H. Persat, R. Eppe, C. Schlötterer & F. Uiblein, 2002. Complex patterns of colonization and refugia revealed for European grayling Thymallus thymallus, based on complete sequencing of the mitochondrial DNA control region. Molecular Ecology 11: 1393–1407.
Weiss, S., I. Knizhin, A. Kirillov & E. Froufe, 2006. Phenotypic and genetic differentiation of two major phylogeographical lineages of arctic grayling Thymallus arcticus in the Lena River, and surrounding Arctic drainages. Biological Journal of the Linnean Society 88: 511–525.
Weiss, S., I. Knizhin, V. Romanov & T. Kopun, 2007. Secondary contact between two divergent lineages of grayling Thymallus in the lower Enisey basin and its taxonomic implications. Journal of Fish Biology 71: 371–386.
Weiss, S. J., T. Kopun & S. Sušnik Bajec, 2013. Assessing natural and disturbed population structure in European grayling Thymallus thymallus: melding phylogeographic, population genetic and jurisdictional perspectives for conservation planning. Journal of Fish Biology 82: 505–521.
Weiss, S., G. Secci-Petretto, A. Antonov & E. Froufe, 2020. Multiple species of grayling (Thymallus sp.) found in sympatry in a remote tributary of the Amur River. Zoologica Scripta 49: 117–128.
Wiens, J. J. & M. J. Donoghue, 2004. Historical biogeography, ecology and species richness. Trends in Ecology & Evolution 19: 639–644.
Winkler, K. A. & S. Weiss, 2008. Eighteen new tetranucleotide microsatellite DNA markers for Coregonus lavaretus cloned from an alpine lake population. Molecular Ecology Resources 8: 1055–1058.
Wong, B. B. M., J. S. Keogh & D. J. McGlashan, 2004. Current and historical patterns of drainage connectivity in eastern Australia inferred from population genetic structuring in a widespread freshwater fish Pseudomugil signifer (Pseudomugilidae). Molecular Ecology 13: 391–401.
Yang, T., W. Meng, R. Zhang, T. Gao, L. Cai, S. Hai & Q. Zhou, 2016. DNA barcoding of fishes in Irtysh River China. Russian Journal of Genetics 52: 969–976.
Yang, X., H. Liu, Z. Ma, Y. Zou, M. Zou, Y. Mao, X. Li, H. Wang, T. Chen, W. Wang & R. Yang, 2019. Chromosomelevel genome assembly of Triplophysa tibetana, a fish adapted to the harsh high altitude environment of the Tibetan Plateau. Molecular Ecology Resources 19: 1027–1036.
Yao, Y. & H. G. Li, 2010. Tectonic geomorphological characteristics for evolution of the Manas Lake. Journal of Arid Land 2: 167–173.
Acknowledgements
Open access funding provided by University of Graz. This work was supported by a grant from the Portuguese Foundation for Science and Technology (SFRH/BD/139069/2018) to G. S., by the Portuguese Strategic Funding UIDB/04423/2020 and UIDP/04423/2020, and by a grant from the Doctoral Program Ecology and Evolution in Changing Environments (EECE) of the University Graz to G. K. E. under the supervision of S. W.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Christian Sturmbauer
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Weiss, S., Grimm, J., Gonçalves, D.V. et al. Comparative genetic analysis of grayling (Thymallus spp. Salmonidae) across the paleohydrologically dynamic river drainages of the Altai-Sayan mountain region. Hydrobiologia 847, 2823–2844 (2020). https://doi.org/10.1007/s10750-020-04273-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-020-04273-3