
Abstract
Improvements in therapies for heart failure with preserved ejection fraction (HFpEF) are crucial for improving patient outcomes and quality of life. Although HFpEF is the predominant heart failure type among older individuals, its prognosis is often poor owing to the lack of effective therapies. The roles of the spleen and bone marrow are often overlooked in the context of HFpEF. Recent studies suggest that the spleen and bone marrow could play key roles in HFpEF, especially in relation to inflammation and immune responses. The bone marrow can increase production of certain immune cells that can migrate to the heart and contribute to disease. The spleen can contribute to immune responses that either protect or exacerbate heart failure. Extramedullary hematopoiesis in the spleen could play a crucial role in HFpEF. Increased metabolic activity in the spleen, immune cell production and mobilization to the heart, and concomitant cytokine production may occur in heart failure. This leads to systemic chronic inflammation, along with an imbalance of immune cells (macrophages) in the heart, resulting in chronic inflammation and progressive fibrosis, potentially leading to decreased cardiac function. The bone marrow and spleen are involved in altered iron metabolism and anemia, which also contribute to HFpEF. This review presents the concept of an interplay between the heart, spleen, and bone marrow in the setting of HFpEF, with a particular focus on extramedullary hematopoiesis in the spleen. The aim of this review is to discern whether the spleen can serve as a new therapeutic target for HFpEF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An estimated 64 million people experience heart failure worldwide [1]. Heart failure is the leading cause of hospitalization among adults, with 1-year mortality reported to be between 10 and 35% in various population-wide registries [2,3,4]. Non-communicable diseases such as obesity, hypertension, and diabetes are major risk factors for heart failure [2, 5]. Heart failure can be grouped into three categories based on the percentage of ejection fraction [6]: heart failure with preserved ejection fraction (HFpEF), heart failure with reduced ejection fraction (HFrEF), and heart failure with midrange ejection fraction (HFmrEF) [6, 7]. HFpEF is defined by an ejection fraction > 50% and is characterized by structural and cellular alterations such as cardiomyocyte hypertrophy, fibrosis, and inflammation, resulting in an inability of the left ventricle to relax properly [7, 8]. HFrEF is defined by an ejection fraction < 40% and is characterized by cardiomyocyte loss, resulting in the development of systolic dysfunction, whereby the left ventricle cannot contract properly [8, 9]. HFmrEF is defined as an ejection fraction of 40–50% [10].
Among patients aged > 65 years with heart failure, almost half have HFpEF [1, 5, 11]. A study conducted to examine biomarkers in heart failure found that biomarkers in HFpEF were associated with markers for inflammation, remodeling, angiogenesis, and hematopoiesis [12]. HFpEF is also associated with elevated levels of pro-inflammatory cytokines [13]. While efficient and specific treatments for HFrEF have been developed, treatment options for HFpEF are lacking [14].
Hematopoiesis is defined as the production of all components of blood and plasma, including cells of the immune system [15]. Hematopoiesis occurs within the hematopoietic system, while bone marrow is the main site for hematopoiesis in adults, and growing evidence points to a role for the spleen in the maintenance and differentiation of hematopoietic stem cells [16]. Extramedullary hematopoiesis refers to blood cell production outside the bone marrow, which can occur in the spleen, liver, adipose tissue, and lymph nodes [17]. The spleen plays an important role in filtering blood and regulating immune responses to various agents and contains large pools of undifferentiated monocytes that can undergo splenic hematopoiesis [18].
Altered iron metabolism and anemia can contribute to HFpEF, and iron deficiency is common in heart failure. The threshold for iron deficiency in patients with heart failure is defined as ferritin of < 100 µg/l or ferritin of 100–300 µg/l if transferrin saturation is < 20% [19,20,21]. This threshold for iron deficiency has been applied in several studies. Jankowska et al. investigated the relationship between iron deficiency and survival in patients with systolic heart failure and reported a prevalence of iron deficiency in patients with heart failure of 37% (in 546 patients) [22]. The PrEP Registry, a prospective observational study conducted by von Haehling et al., assessed the prevalence and clinical impact of iron deficiency and anemia in heart failure patients in Germany and found a prevalence of 42.5% (in 1198 patients) [23]. Iron deficiency can lead to anemia, which is caused by a decrease in iron levels characterized by decreased levels of hematopoiesis in red blood cells (hypochromia) and diminished mean corpuscular volume (microcytosis) [24]. Most iron in the body is bound to hemoglobin in erythrocytes, with the rest being stored in macrophages and hepatocytes [25]. Besides the bone marrow and liver, the spleen participates in iron metabolism, and macrophages in the red pulp of the spleen recycle iron from senescent red blood cells [25]. Anemia often develops in patients with HFpEF. Its prevalence has been reported over a variety of ranges: 21–68% in hospitalized patients, 19–27% in participants of randomized controlled trials, and 30–33% in patients being treated on an outpatient basis [26]. Anemia is classified as hemoglobin < 13.0 g/dl in men and < 12.0 g/dl in women [27]. The origin of anemia in heart failure is multifactorial and includes important factors such as renal dysfunction, hemodilution, chronic inflammation, certain therapeutics (for example, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or beta blockers), bone marrow dysfunction, resistance to erythropoietin, and deficiencies of vitamin B12, folic acid, and iron [28,29,30].
The role of inflammation in both the onset and progression of heart failure is also of great interest. Most major immune cell types in the steady state are found in the heart, including monocytes, macrophages, T cells, neutrophils, B cells, dendritic cells, natural killer cells, and mast cells [31]. The spleen, being the body’s largest secondary immune organ, hosts a wide range of immunological functions besides its roles in hematopoiesis and red blood cell clearance [18].
This review aims to assess the current status of the role played by extramedullary hematopoiesis in the interplay between the heart, spleen, and bone marrow in the context of heart failure, especially HFpEF. Iron metabolism and anemia are also discussed in view of heart failure. Additionally, this review examines hematopoiesis other than that in the bone marrow, specifically extramedullary emergency hematopoiesis in the spleen, which may be involved in the pathogenesis and progression of HFpEF.
Medullary hematopoiesis and heart failure
There is increasing evidence of a link between medullary hematopoiesis (occurring in the bone marrow) and heart failure. Clinical studies have shown that levels of bone marrow-derived circulating endothelial progenitor cells are associated with cardiovascular risk factors, including increased age, diabetes, smoking, and hypertension [32].
Clonal hematopoiesis
Clonal hematopoiesis is a pre-leukemic condition in which hematopoietic stem cells with acquired somatic DNA mutations proliferate in the bone marrow, and its frequency has been reported to increase with age [33]. Previously, the pathological significance of clonal hematopoiesis beyond increasing the risk of hematological malignancy was not well understood, but recent epidemiological analyses and animal experiments have revealed that it is a novel risk factor for cardiovascular diseases [34,35,36]. Carriers of hematopoiesis of indeterminate potential (CHIP) were shown to have a 1.9-fold increased risk of heart disease compared with non-carriers, and somatic mutations in DNA methyltransferase DNMT3A, DNA demethylase TET2, additional sex combs-like 1 ASXL1, and Janus kinase 2 JAK2 have been associated with ischemic heart disease [34]. Furthermore, it has been reported that clonal hematopoiesis may be involved in the pathogenesis of heart failure of ischemic origin [37]. CHIP driver genes TET2 and DNMT3A have been shown to be significantly associated with the progression and poor prognosis of ischemic heart failure [37]. Subsequent analyses have reported, from an epidemiological perspective, that clonal hematopoiesis is significantly associated with the progression and poor prognosis of HFrEF, regardless of the etiology [38]. In addition, mutations in TET2 or DNMT3A have demonstrated a profoundly increased mortality driven by the progression of heart failure [37]. Furthermore, TET2 CHIP has been identified as an independent risk factor for HFpEF, independent of conventional cardiovascular risk factors and coronary artery disease [39]. The mechanism by which clonal hematopoiesis of TET2 and DNMT3A exacerbates atherosclerosis and heart failure is proposed to be that mutated macrophages become pro-inflammatory, leading to the growth of arterial plaques and myocardial damage [36, 40,41,42,43]. Moreover, clonal hematopoiesis is frequently observed not only in the elderly but also in relatively younger patients who have undergone cancer treatment. In addition, therapy-related clonal hematopoiesis is a condition found in cancer survivors that may contribute to an increased risk of cardiovascular events [41]. In these cases, clonal hematopoiesis frequently detects DNA damage response genes such as protein phosphatase, Mg2+/Mn2+ dependent 1D, PPM1D; tumor protein p53, TP53; ataxia-telangiectasia mutated, ATM; and checkpoint serine-threonine kinase 2, CHEK2 [44]. Recent experimental studies have reported that clonal hematopoiesis due to PPM1D and TP53 genes exacerbates hypertensive heart failure and doxorubicin cardiomyopathy, respectively [39, 41, 45]. While previous reports on heart failure primarily focused on HFrEF, very recent studies have shown that clonal hematopoiesis is also associated with the pathology of HFpEF, garnering significant attention [40]. For instance, clonal hematopoiesis is thought to contribute to the formation of a distinct population of blood cells with acquired DNA mutations that are associated with an increased risk of heart failure, especially HFpEF [34, 37, 39, 40]. A study investigated the significance of clonal hematopoiesis in patients with HFpEF versus those without HFpEF [40]. The study found that TET2-mediated clonal hematopoiesis was enriched among patients with HFpEF, and that these patients had worse heart function and prognosis, especially those aged ≥ 70 years [40]. In the same study, the authors used a Tet2 clonal hematopoiesis model mouse induced to develop HFpEF via high-fat diet and Nω-nitro-L-arginine methyl ester (L-NAME) stimulation. They reported significant diastolic dysfunction in the Tet2 group, as demonstrated by echocardiography and catheter examinations [40]. Additionally, a large-scale epidemiological study also reported that clonal hematopoiesis is associated with HF, with a stronger association with HFpEF [39]. Additional studies in mice have revealed evidence for immune dysregulation in HFpEF and that genetic mutations associated with CHIP induce skewing of immune cells to a pro-inflammatory phenotype in vivo and in vitro [46, 47]. Inactivating Tet2 and Dnmt3 in mice has promoted angiotensin-II-induced cardiac dysfunction [46]. Furthermore, Tet2 deficiency in hematopoietic cells in mice is associated with greater cardiac dysfunction resulting from elevated signaling of interleukin-1β [47]. Thus, clonal hematopoiesis, including medullary hematopoiesis, can be considered an important factor modifying heart failure, and the development of diagnostic and therapeutic approaches focusing on these aspects is highly anticipated.
Extramedullary hematopoiesis in the spleen and heart failure
Extramedullary hematopoiesis is the term used to describe the process of hematopoiesis that occurs in organs other than the bone marrow [17, 48]. For example, during immune responses, the spleen or liver can serve as sites for extramedullary hematopoiesis [48]. It is important to point out that studies conducted with humans are lacking. Therefore, the data provided in this section are derived from research carried out on animals. Evidence from a mouse model indicates that extramedullary hematopoiesis in the spleen contributes to the risk of heart failure [48]. Another mouse model study showed that deletion of a G-protein-coupled receptor ALX results in inflammation, along with an increase of pro-inflammatory Ly6Chi C–C chemokine receptor type 2+ macrophages in the spleen and heart at a steady state, leading to an inflamed splenocardiac axis [49]. The authors suggested that ALX receptor deficiency may provide an experimental model that includes cellular and molecular mechanisms observed in HFpEF. Research on the impact of extramedullary hematopoiesis in the spleen on the risk of heart failure in humans is needed. Physiologic extramedullary hematopoiesis occurs in fetuses but not in adults [17]. However, when triggered by stress or disease, the spleen undergoes extramedullary hematopoiesis in adults [48]. Extramedullary hematopoiesis in the spleen can be a response to hematopoietic stress caused by microbial infections and diseases such as myelofibrosis [50], malaria [51], atherosclerosis [15], and osteopetrosis [52]. A recent study examined the effect of psychosocial stress on mice exposed to mild social defeat stress, with the authors observing severe extramedullary hematopoiesis in the spleen in mice exposed to social defeat stress [53].
Spleen and heart failure
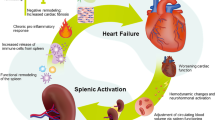
The links between the spleen and heart in heart failure, termed the cardiosplenic axis, were described in detail in a recent review by our group [54]. Of note, the causal directionality of the relationship between spleen size and heart failure may not always be apparent [54]. An interplay between chemical messengers sent out by the heart and received by the spleen may promote heart failure (Fig. 1). For example, in the infarcted heart, infiltrating neutrophils release alarmins that can activate splenic cells to produce immunoglobulin G; this accumulates in the atherosclerotic plaque, forming immune cell-immunoglobulin G complexes that promote inflammation [55, 56]. Additionally, an interaction exists among the sympathetic nervous system, bone marrow, and spleen in heart failure. The sympathetic nervous system is stimulated after myocardial infarction, which relays signals back to the bone marrow. This results in the release of hematopoietic stem cells, which travel to the spleen and undergo extramedullary hematopoiesis [57]. Monocytes produced via extramedullary hematopoiesis stimulated by the sympathetic nervous system go on to infiltrate atherosclerotic lesions, increasing the susceptibility of the lesions to rupture and leading to an increased risk of secondary myocardial infarction [57]. The spleen performs highly important hematological and immunological functions, and the structure and function of the spleen have been described elsewhere [54].

Model of the interplay of the heart, bone marrow, and spleen relating to heart failure, from the perspective of spleen-based extramedullary hematopoiesis
A recent review looked at the role of splanchnic circulation in the pathogenesis of heart failure, along with potential novel treatment options for redistributing blood volume in patients with heart failure, especially HFpEF [58]. The highly vascularized organs of the splanchnic compartment, which include the liver, spleen, and mesenteric vasculature, hold the majority of the body’s intravascular blood volume. Excess fluid redistribution from this compartment leads to increased cardiac filling pressures. This inappropriate regulation of blood volume distribution is suggested to be a factor contributing to the exercise intolerance seen in HFpEF [58]. The role of the sympathetic nervous system in modulating the capacitance and compliance of the splanchnic vascular bed via modulation of the greater splanchnic nerve, and greater splanchnic nerve ablation for volume management, have been proposed as a potential therapeutic intervention to increase unstressed blood volume. Studies have shown that right-sided greater splanchnic nerve ablation was safe and improved mostly subjective clinical metrics such as N-terminal pro-brain natriuretic peptide, health status, and 6-min walk distance (6MWD) in patients with HFpEF over 12 months [59, 60]. These findings suggest that splanchnic nerve modulation provides a strong foundation for research into this potential therapeutic target in HFpEF.
The outcome in patients who do not have a spleen or who have undergone a splenectomy is often poor. Although other organs such as the liver and bone marrow can take over most of the spleen’s functions, patients without a spleen have an increased risk of infections, given that the spleen plays a significant role in fighting infections [61]. A major contributor to poor outcomes after splenectomy is an increase in infections and infectious complications [62, 63]. Surgical removal of the spleen in humans can result in elevated low-density lipoprotein cholesterol, altered lipid values, and atherosclerosis [63, 64]. A 2016 study examining long-term outcomes after pediatric splenectomy [62] reported various findings, including a very low risk of portal vein thrombosis, the association of sepsis with underlying immunodeficiency, and a low recurrence rate of hematologic disorders. A study using a rat model found splenectomy to improve portal hypertension in both cirrhotic and non-cirrhotic models, while hypertension related to anemia and thrombocytopenia was improved in the non-cirrhotic model [65]. Isolated congenital asplenia, which is an extremely rare condition, has been described in a case report [66]. The patient was diagnosed with reactive thrombocytosis related to hyposplenism, with the authors suggesting vaccination against the main encapsulated bacteria in such cases, given the high mortality associated with such infections [66]. A 2014 study included 8149 USA veterans who underwent splenectomy with up to 27 years of follow-up. Those who underwent splenectomy had increased risks of infections, thromboembolism, coronary artery disease, and malignancies that persisted over 10 years after splenectomy [67]. Another study found that World War II servicemen who had undergone splenectomy because of trauma were 1.9 times more likely to die of ischemic heart disease [68]. While a splenectomy does not decrease the risk of heart failure, it may increase the risk of cardiac complications. Conversely, hypersplenism may be a cardiovascular risk factor for patients with heart failure [69]. Red blood cell destruction is a manifestation of hypersplenism and may contribute to the increased production of reactive oxygen species in heart failure progression. Table 1 shows clinical data that support the critical importance of the spleen for heart failure progression.
Iron metabolism in heart failure
Heart failure is frequently associated with iron deficiency with or without anemia [70]. Iron is essential for the optimal functioning and survival of living organisms. Iron deficiency leads to mitochondrial dysfunction, aberrant enzyme activity, abnormal transport of structural proteins, and apoptosis [71]. Moreover, the ability of organs and tissues to function properly is impaired, along with decreased exercise capacity, work functioning, and cognitive performance, and increased morbidity and mortality [71].
Two types of iron deficiency have been proposed: absolute iron deficiency and functional iron deficiency. Absolute iron deficiency is defined by severely reduced or absent iron stores in bone marrow, liver, and spleen [71]. Functional iron deficiency is defined by normal or increased total body iron stores that are unavailable for incorporation into erythroid precursors for erythropoiesis owing to increased levels of hepcidin [71]. Patients with heart failure and iron deficiency have lower levels of iron in the spleen and liver [72]. The spleen plays an important role in the circulatory system, i.e., removal of aged or damaged red blood cells. Macrophages that reside in the red pulp of the spleen, characterized by the expression of CD163, mediate the turnover of aging red blood cells [25]. Furthermore, the spleen participates in red blood cell turnover by inducing hemolysis [25]. Aging red blood cells undergoing hemolysis in the spleen release hemoglobin to the environment; this may bind to the hemoglobin haptoglobin scavenger receptor, CD163, which is highly expressed in the spleen [73]. These findings provide evidence for the important role of the spleen in efficient iron recycling from senescent red blood cells.
Myocardial iron deficiency in heart failure is accompanied by decreased mitochondrial functions, diminished activity of citric acid cycle enzymes, and reduced expression of reactive oxygen species protective enzymes, suggesting that myocardial iron deficiency may contribute to myocardial dysfunction in heart failure [74]. Patients with heart failure and iron deficiency showed lower levels of iron in the spleen and liver, with a trend toward lower iron content in the heart’s septum [71]. Furthermore, iron levels in the left ventricle were significantly decreased in failing hearts compared with those in non-failing hearts [74]. These effects of iron deficiency can be reversed with therapy. After intravenous iron therapy, iron levels increased in the left ventricle and spleen [72, 75].
Anemia and heart failure
Iron deficiency is the most common cause of anemia in patients with heart failure [76]. Mortality is higher in patients with heart failure and anemia than in patients with heart failure without anemia, even after adjustment for potential confounders [77]. A study in a Swedish heart failure registry explored the relationships between anemia and heart failure across the ejection fraction spectrum [78]. Anemia prevalence was the highest in HFpEF; anemia was independently associated with a similar risk of death regardless of ejection fraction but with a higher risk of heart failure hospitalization in HFpEF and HFmrEF than in HFrEF [78].
Renal dysfunction and chronic kidney disease are common complications of heart failure. The interaction among anemia, congestive heart failure, and chronic kidney insufficiency has been described as a vicious cycle termed cardio-renal anemia syndrome [79]. In chronic kidney disease and other conditions with impaired renal function, the production of erythropoietin, which stimulates the production of blood cells, is reduced, causing anemia [80]. Production of erythropoietin is regulated by hypoxia-inducible factor (HIF) [81]. Inhibitors of HIF prolyl-hydroxylase (HIF-PH) have recently been used to treat renal anemia and have improved anemia caused by chronic kidney disease and other conditions [80, 82]. Findings from a recent study by Nakamura et al. suggest that HIF-PH inhibitors may be safe and effective for treating renal anemia in patients with heart failure [83]. The FAIR-HF trial showed that treatment with intravenous ferric carboxymaltose in patients with chronic heart failure and iron deficiency with or without anemia improved symptoms and quality of life [84]. Furthermore, intravenous iron therapy in patients with heart failure with or without anemia demonstrated improvement in subjective symptoms, quality of life, and the 6MWD test [84]. A meta-analysis of data from several clinical trials indicated that treatment with intravenous ferric carboxymaltose is associated with a reduction in recurrent hospitalizations in patients with heart failure and iron deficiency [85]. Treatment of patients with acute non-compensated heart failure and iron deficiency with ferric carboxymaltose was safe and reduced the risk of hospitalization for heart failure [86]. In addition, patients with chronic heart failure treated with intravenous iron alone reported improvements in their subjective symptoms and an improved 6MWD with increased hemoglobin [87]. Sodium-glucose cotransporter 2 (SGLT2) inhibition with empagliflozin may stimulate erythropoiesis and improve functional iron deficiency in patients with type 2 diabetes and heart failure [88]. The DAPA-HF trial examined the effect of SGLT2 inhibitors on outcomes in patients exhibiting a wide range of HFrEF. The study demonstrated that the impact of dapagliflozin on anemia reversal remained steady, irrespective of whether iron deficiency was present or absent initially. The study indicated a potential therapeutic synergy between iron supplementation and SGLT2 inhibition, which could be beneficial for some patients with HFrEF to prevent iron deficiency and to treat anemia [89]. In addition, the 2022 American College of Cardiology/American Heart Association/Heart Failure Association of America Guideline for the Management of Heart Failure [21] recommends SGLT2 inhibition for patients because it has been shown to decrease the risk of hospitalization for heart failure and cardiovascular mortality in patients with heart failure, regardless of whether they have type 2 diabetes. This benefit applies for both patients with HFrEF and those with HFpEF [90]. However, a recent review pointed out that SGLT2 inhibitors have been shown to decrease hepcidin and ferritin and increase transferrin receptor protein, which is suggestive of worsening iron deficiency [91]. That review advocates for additional studies of the efficacy and safety of concurrent use of SGLT2 inhibitors and intravenous iron in patients with heart failure.
Links between inflammation and heart failure
The inflammasome (a part of the innate immune system) has been investigated in heart failure and has been shown to play a role in pathological changes in the heart, such as hypertrophy, fibrosis, and cell death [92, 93]. Multiple studies have highlighted the importance of sterile inflammation in heart failure. Sterile inflammation occurs in the absence of infection and involves the secretion of inflammatory cytokines and the recruitment of innate immune cells, such as neutrophils and monocytes/macrophages [94, 95]. Inflammation leads to an increased release of hepcidin from the liver, which downregulates ferroportin, resulting in (1) reduced transport of dietary iron intake from the inside of mucosal cells to the small intestine to the bloodstream [96] and (2) reduced release of recycled iron from the macrophages in the spleen and liver to the bloodstream, which in turn may lead to iron deficiency [25].
Immune dysregulation plays a role in heart failure via extramedullary hematopoiesis occurring in the spleen. A study in mice has shown that the spleen is a major site of macrophage production after myocardial infarction [97]. The authors screened spleen mRNA levels after myocardial infarction and found a marked increase in the expression of interleukin-1β [97]. Of note, pro-inflammatory cytokines such as tumor necrosis factor-α and interleukin-6 have been found to have a direct effect on the bone marrow and may be involved in the mechanism of anemia in chronic disease [98]. Ischemic injury to the heart leads to the replacement of the cardiac resident macrophages in the infarct zone with monocyte-derived macrophages from the bone marrow and spleen, which may result in a pro-inflammatory environment [99]. However, a study provided evidence that splenic leukocytes and macrophages promote left ventricle healing by generating specialized proresolving mediators, such as lipoxins, resolvins, protectin, and maresin, which are enzymatically produced during the resolution of inflammation [100, 101]. Heart failure induces monocytopoiesis in the bone marrow and in the spleen [57, 102]. The PREFER-HF study will comprehensively evaluate the relationship between clinical characteristics, genomic, proteomic, and metabolomic data, along with imaging information and clinical outcomes in a US cohort receiving ambulatory care [103]. Data are expected to be reported in approximately 5 years.
Immune cell mobilization in heart failure
The spleen may compensate for decreased bone marrow function associated with heart failure by increasing its own function [57]. A study on the effects of social stress in mice revealed the possibility that splenic erythropoiesis may partially alleviate the anemia associated with stressful situations (inflammation) [104]. The phenotype of cardiac resident macrophages differs from blood-derived macrophages from the spleen [98]. Cardiac macrophages have an M2-like gene expression signature, suggesting that these macrophages play an immunomodulatory function in the heart [105]. Cardiac resident macrophages are protective for the heart, while spleen-derived macrophages help heal the injured myocardium and promote pathology [106]. Elevated leukocyte counts have been shown in patients with heart failure after myocardial infarction [100]. In addition, the spleen has been shown to provide a steady flow of leukocytes to the left ventricle after myocardial infarction [100]. A study looked at leukocyte trafficking and specialized proresolving mediators in the spleen and in the left ventricle after myocardial infarction [100]. The study’s findings suggested that leukocytes mobilize from the spleen to the heart and generate lipids that aid in resolving the inflammation after myocardial infarction [100]. Recent findings have described an iron-dependent form of cell death termed ferroptosis in cardiac injury resulting from both chemotherapy and ischemia/reperfusion [107]. Ferroptosis is distinct from both apoptosis and necroptosis and can be halted or reversed with iron chelation [107, 108]. Neutrophils are thought to impair myocardial repair and contribute to heart failure progression via the generation of damaging cytokines and reactive oxygen species and the release of enzymes stored in secretory granules [109]. Neutrophils may have cardioprotective properties such that the generation of reactive oxygen species by neutrophils may stimulate the differentiation of reparative macrophages [110].
Clinical implications and future directions
Further exploration of the interplay between the heart, spleen, and bone marrow, from the viewpoint of extramedullary hematopoiesis, is warranted. Many of the findings have been developed using mouse models and may not be representative of patients with cardiac diseases. In fact, the absence of a strong HFpEF in vivo model restricts our understanding of the pathophysiology of HFpEF. No existing in vivo model encapsulates all the hemodynamic features of HFpEF [111, 112]. This presents a significant challenge in our understanding and research of this condition. Animal models of HFpEF are under active investigation. A recent study created several mouse models of HFpEF by combining three independent pathological stressors (increased calcium ion influx, high-fat diet, and L-NAME). The study showed that histone deacetylase 6 (Hdac6) deletion in mice slows HFpEF progression, and that a specific HDAC6 inhibitor, TYA-018, reverses existing cardiac hypertrophy and diastolic dysfunction in mouse models of HFpEF [113], suggesting that HDAC6 inhibition may be a therapeutic approach for HFpEF.
In humans, two clinical trials have explored the effect of the SGLT2 inhibitor empagliflozin in HFpEF [114,115,116]. The placebo-controlled EMPEROR-Preserved trial evaluated the effect of empagliflozin on a primary outcome of a composite of cardiovascular death or hospitalization for heart failure, reporting that empagliflozin reduced the risk of the primary outcome in patients with HFpEF, regardless of the presence or absence of diabetes [116]. The EMPERIAL-Preserved trial evaluated the effects of empagliflozin on exercise ability and patient-reported outcomes in patients with HFpEF, with or without diabetes [115]. While the effect of empagliflozin on the primary outcome—6MWD—was neutral, the authors noted that empagliflozin was well tolerated in HFpEF patients, both with and without diabetes.
Overall, further studies are needed to better understand the effects of extramedullary hematopoiesis on heart disease, and further research in animal models of HFpEF and of therapeutic approaches in patients with HFpEF is welcome. Table 2 provides proposed hypotheses from the viewpoint of extramedullary hematopoiesis in the spleen in heart failure.
Conclusions
Medullary hematopoiesis plays a role in heart failure. Bone marrow-derived circulating endothelial progenitor cells are associated with cardiovascular risk factors, including increased age, diabetes, smoking, and hypertension. Clonal hematopoiesis, which occurs in the bone marrow, especially in older adults, is consequently involved in the development of cardiovascular diseases, including heart failure. This is owing to the abnormal immune cells produced. The spleen serves as an important source of immune system cells, and immune dysregulation plays a role in heart failure through extramedullary hematopoiesis occurring in the spleen. In heart failure, emergency hematopoiesis occurs in the spleen, i.e., immune cells are also produced (monocytes, macrophages, neutrophils), which are mobilized to the heart and the rest of the body. Therefore, we speculate that extramedullary hematopoiesis may be a key factor in the development of heart failure, especially HFpEF. Extramedullary hematopoiesis in the spleen is a compensatory change in the pathogenesis of heart failure but may also be a mechanism for the progression and exacerbation of chronic heart failure (especially HFpEF). This results from the repeated increase in metabolic activity in the spleen, immune cell production, mobilization to the heart, and concomitant cytokine production. Ultimately, this leads to systemic chronic inflammation, as well as to an imbalance of immune cells (macrophages) in the heart, resulting in chronic inflammation and progressive fibrosis, potentially causing decreased cardiac function.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
GBD Disease and Injury Incidence and Prevalence Collaborators (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 392(10159):1789–1858. https://doi.org/10.1016/S0140-6736(18)32279-7
Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN, Gheorghiade M (2014) The global health and economic burden of hospitalizations for heart failure: Lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 63(12):1123–1133. https://doi.org/10.1016/j.jacc.2013.11.053
Lund LH, Carrero JJ, Farahmand B, Henriksson KM, Jonsson A, Jernberg T, Dahlstrom U (2017) Association between enrolment in a heart failure quality registry and subsequent mortality-a nationwide cohort study. Eur J Heart Fail 19(9):1107–1116. https://doi.org/10.1002/ejhf.762
Crespo-Leiro MG, Metra M, Lund LH et al (2018) Advanced heart failure: A position statement of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 20(11):1505–1535. https://doi.org/10.1002/ejhf.1236
Wang Z, Yang T, Fu H (2021) Prevalence of diabetes and hypertension and their interaction effects on cardio-cerebrovascular diseases: A cross-sectional study. BMC Public Health 21(1):1224. https://doi.org/10.1186/s12889-021-11122-y
Borlaug BA, Redfield MM (2011) Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 123(18):2006–2013. https://doi.org/10.1161/CIRCULATIONAHA.110.954388. discussion 2014
Simmonds SJ, Cuijpers I, Heymans S, Jones EAV (2020) Cellular and molecular differences between HFpEF and HFrEF: A step ahead in an improved pathological understanding. Cells 9(1):242. https://doi.org/10.3390/cells9010242
Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT, Granzier H, Hummel SL, Kass DA, Redfield MM, Sam F, Wang TJ, Desvigne-Nickens P, Adhikari BB (2020) Research priorities for heart failure with preserved ejection fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 141(12):1001–1026. https://doi.org/10.1161/CIRCULATIONAHA.119.041886
Miller WL, Grill DE, Borlaug BA (2013) Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: pulmonary hypertension and heart failure. JACC Heart Fail 1(4):290–299. https://doi.org/10.1016/j.jchf.2013.05.001
Lam CS, Solomon SD (2014) The middle child in heart failure: Heart failure with mid-range ejection fraction (40–50%). Eur J Heart Fail 16(10):1049–1055. https://doi.org/10.1002/ejhf.159
Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM (2012) Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail 5(6):720–726. https://doi.org/10.1161/CIRCHEARTFAILURE.111.966366
Tromp J, Khan MAF, Mentz RJ, O’Connor CM, Metra M, Dittrich HC, Ponikowski P, Teerlink JR, Cotter G, Davison B, Cleland JGF, Givertz MM, Bloomfield DM, Van Veldhuisen DJ, Hillege HL, Voors AA, van der Meer P (2017) Biomarker profiles of acute heart failure patients with a mid-range ejection fraction. JACC Heart Fail 5(7):507–517. https://doi.org/10.1016/j.jchf.2017.04.007
Adamo L, Rocha-Resende C, Prabhu SD, Mann DL (2021) Publisher correction: Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 18(10):735. https://doi.org/10.1038/s41569-021-00534-3
Reddy YNV, Rikhi A, Obokata M, Shah SJ, Lewis GD, AbouEzzedine OF, Dunlay S, McNulty S, Chakraborty H, Stevenson LW, Redfield MM, Borlaug BA (2020) Quality of life in heart failure with preserved ejection fraction: Importance of obesity, functional capacity, and physical inactivity. Eur J Heart Fail 22(6):1009–1018. https://doi.org/10.1002/ejhf.1788
Fernandez-Garcia V, Gonzalez-Ramos S, Martin-Sanz P, Castrillo A, Bosca L (2020) Contribution of extramedullary hematopoiesis to atherosclerosis. The spleen as a neglected hub of inflammatory cells. Front Immunol 11:586527. https://doi.org/10.3389/fimmu.2020.586527
O’Neill HC, Lim HK, Periasamy P, Kumarappan L, Tan JKH, O’Neill TJ (2019) Transplanted spleen stromal cells with osteogenic potential support ectopic myelopoiesis. PLoS ONE 14(10):e0223416. https://doi.org/10.1371/journal.pone.0223416
Kim CH (2010) Homeostatic and pathogenic extramedullary hematopoiesis. J Blood Med 1:13–19. https://doi.org/10.2147/JBM.S7224
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ (2009) Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325(5940):612–616. https://doi.org/10.1126/science.1175202
von Haehling S, Ebner N, Evertz R, Ponikowski P, Anker SD (2019) Iron deficiency in heart failure: An overview. JACC Heart Fail 7(1):36–46. https://doi.org/10.1016/j.jchf.2018.07.015
Ponikowski P, Voors AA, Anker SD et al (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37(27):2129–2200. https://doi.org/10.1093/eurheartj/ehw128
Heidenreich PA, Bozkurt B, Aguilar D et al (2022) 2022 AHA/ACC/HFSA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 79(17):e263–e421. https://doi.org/10.1016/j.jacc.2021.12.012
Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, Borodulin-Nadzieja L, Banasiak W, Polonski L, Filippatos G, McMurray JJ, Anker SD, Ponikowski P (2010) Iron deficiency: An ominous sign in patients with systolic chronic heart failure. Eur Heart J 31(15):1872–1880. https://doi.org/10.1093/eurheartj/ehq158
von Haehling S, Gremmler U, Krumm M, Mibach F, Schon N, Taggeselle J, Dahm JB, Angermann CE (2017) Prevalence and clinical impact of iron deficiency and anaemia among outpatients with chronic heart failure: The PrEP Registry. Clin Res Cardiol 106(6):436–443. https://doi.org/10.1007/s00392-016-1073-y
Camaschella C (2017) New insights into iron deficiency and iron deficiency anemia. Blood Rev 31(4):225–233. https://doi.org/10.1016/j.blre.2017.02.004
Klei TRL, Dalimot J, Nota B, Veldthuis M, Mul FPJ, Rademakers T, Hoogenboezem M, Nagelkerke SQ, van IJcken WFJ, Oole E, Svendsen P, Moestrup SK, van Alphen FPJ, Meijer AB, Kuijpers TW, van Zwieten R, van Bruggen R (2020) Hemolysis in the spleen drives erythrocyte turnover. Blood 136(14):1579–1589. https://doi.org/10.1182/blood.2020005351
Triposkiadis F, Giamouzis G, Parissis J, Starling RC, Boudoulas H, Skoularigis J, Butler J, Filippatos G (2016) Reframing the association and significance of co-morbidities in heart failure. Eur J Heart Fail 18(7):744–758. https://doi.org/10.1002/ejhf.600
Cappellini MD, Motta I (2015) Anemia in clinical practice-definition and classification: Does hemoglobin change with aging? Semin Hematol 52(4):261–269. https://doi.org/10.1053/j.seminhematol.2015.07.006
Komajda M, Anker SD, Charlesworth A, Okonko D, Metra M, Di Lenarda A, Remme W, Moullet C, Swedberg K, Cleland JG, Poole-Wilson PA (2006) The impact of new onset anaemia on morbidity and mortality in chronic heart failure: Results from COMET. Eur Heart J 27(12):1440–1446. https://doi.org/10.1093/eurheartj/ehl012
Ishani A, Weinhandl E, Zhao Z, Gilbertson DT, Collins AJ, Yusuf S, Herzog CA (2005) Angiotensin-converting enzyme inhibitor as a risk factor for the development of anemia, and the impact of incident anemia on mortality in patients with left ventricular dysfunction. J Am Coll Cardiol 45(3):391–399. https://doi.org/10.1016/j.jacc.2004.10.038
Anand IS, Kuskowski MA, Rector TS, Florea VG, Glazer RD, Hester A, Chiang YT, Aknay N, Maggioni AP, Opasich C, Latini R, Cohn JN (2005) Anemia and change in hemoglobin over time related to mortality and morbidity in patients with chronic heart failure: Results from Val-HeFT. Circulation 112(8):1121–1127. https://doi.org/10.1161/CIRCULATIONAHA.104.512988
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD (2016) Revisiting cardiac cellular composition. Circ Res 118(3):400–409. https://doi.org/10.1161/CIRCRESAHA.115.307778
Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348(7):593–600. https://doi.org/10.1056/NEJMoa022287
Shlush LI (2018) Age-related clonal hematopoiesis. Blood 131(5):496–504. https://doi.org/10.1182/blood-2017-07-746453
Jaiswal S, Natarajan P, Silver AJ et al (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377(2):111–121. https://doi.org/10.1056/NEJMoa1701719
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andrés V, Hirschi KK, Martin KA, Walsh K (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355(6327):842–847. https://doi.org/10.1126/science.aag1381
Yura Y, Sano S, Walsh K (2020) Clonal hematopoiesis: A new step linking inflammation to heart failure. JACC Basic Transl Sci 5(2):196–207. https://doi.org/10.1016/j.jacbts.2019.08.006
Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brune B, Wagner S, Serve H, Hoffmann J, Seeger F, Dimmeler S, Zeiher AM, Rieger MA (2019) Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol 4(1):25–33. https://doi.org/10.1001/jamacardio.2018.3965
Pascual-Figal DA, Bayes-Genis A, Díez-Díez M, Hernández-Vicente Á, Vázquez-Andrés D, de la Barrera J, Vazquez E, Quintas A, Zuriaga MA, Asensio-López MC, Dopazo A, Sánchez-Cabo F, Fuster JJ (2021) Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J Am Coll Cardiol 77(14):1747–1759. https://doi.org/10.1016/j.jacc.2021.02.028
Schuermans A, Honigberg MC, Raffield LM, Yu B, Roberts MB, Kooperberg C, Desai P, Carson AP, Shah AM, Ballantyne CM, Bick AG, Natarajan P, Manson JE, Whitsel EA, Eaton CB, Reiner AP (2024) Clonal hematopoiesis and incident heart failure with preserved ejection fraction. JAMA Netw Open 7(1):e2353244. https://doi.org/10.1001/jamanetworkopen.2023.53244
Cochran JD, Yura Y, Thel MC et al (2023) Clonal hematopoiesis in clinical and experimental heart failure with preserved ejection fraction. Circulation 148(15):1165–1178. https://doi.org/10.1161/circulationaha.123.064170
Yura Y, Cochran JD, Walsh K (2022) Therapy-related clonal hematopoiesis: A new link between cancer and cardiovascular disease. Heart Fail Clin 18(3):349–359. https://doi.org/10.1016/j.hfc.2022.02.010
Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, Kour A, Evans MA, Zuriaga MA, Hirschi KK, Fuster JJ, Pietras EM, Walsh K (2020) Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5(6). https://doi.org/10.1172/jci.insight.135204
Shumliakivska M, Luxán G, Hemmerling I, Dimmeler S et al (2024) DNMT3A clonal hematopoiesis-driver mutations induce cardiac fibrosis by paracrine activation of fibroblasts. Nat Commun 15(1):606. https://doi.org/10.1038/s41467-023-43003-w
Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J, Arcila ME, Ladanyi M, Tallman MS, Levine RL, Berger MF (2017) Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21(3):374-382.e374. https://doi.org/10.1016/j.stem.2017.07.010
Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, Kour A, Yura Y, Doviak H, Walsh K (2021) TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight 6(13):e146076. https://doi.org/10.1172/jci.insight.146076
Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K (2018) CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res 123(3):335–341. https://doi.org/10.1161/CIRCRESAHA.118.313225
Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, Fuster JJ, Walsh K (2018) Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol 71(8):875–886. https://doi.org/10.1016/j.jacc.2017.12.037
Wang L, Yang M, Arias A, Song L, Li F, Tian F, Qin M, Yukht A, Williamson IK, Shah PK, Sharifi BG (2015) Splenocytes seed bone marrow of myeloablated mice: implication for atherosclerosis. PLoS ONE 10(6):e0125961. https://doi.org/10.1371/journal.pone.0125961
Tourki B, Kain V, Shaikh SR, Leroy X, Serhan CN, Halade GV (2020) Deficit of resolution receptor magnifies inflammatory leukocyte directed cardiorenal and endothelial dysfunction with signs of cardiomyopathy of obesity. Faseb j 34(8):10560–10573. https://doi.org/10.1096/fj.202000495RR
Ojeda-Uribe M, Morel O, Ungureanu C, Desterke C, Le Bousse-Kerdiles MC, Boulahdour H (2016) Assessment of sites of marrow and extramedullary hematopoiesis by hybrid imaging in primary myelofibrosis patients. Cancer Med 5(9):2378–2384. https://doi.org/10.1002/cam4.835
Mungyer G, Poels LG, Jerusalem C, Jerusalem R (1983) Plasmodium berghei: Influence on granulopoiesis and macrophage production in BALB/c mice. Exp Parasitol 56(2):266–276. https://doi.org/10.1016/0014-4894(83)90072-3
Freedman MH, Saunders EF (1981) Hematopoiesis in the human spleen. Am J Hematol 11(3):271–275. https://doi.org/10.1002/ajh.2830110307
Toyoda A, Kawakami K, Amano Y, Nishizawa H, Nakamura SI, Kawase T, Yoshida Y, Suzuki H, Tsukahara T (2023) Impacts of subchronic and mild social defeat stress on plasma putrefactive metabolites and cardiovascular structure in male mice. Int J Mol Sci 24(2):1237. https://doi.org/10.3390/ijms24021237
Hiraiwa H, Okumura T, Murohara T (2022) The cardiosplenic axis: The prognostic role of the spleen in heart failure. Heart Fail Rev 27(6):2005–2015. https://doi.org/10.1007/s10741-022-10248-4
Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, Lal H, Schroder K, Hanaoka BY, Raman C, Grant MB, Hudson JE, Smyth SS, Porrello ER, Murphy AJ, Nagareddy PR (2020) Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation 141(13):1080–1094. https://doi.org/10.1161/CIRCULATIONAHA.119.043833
Kyaw T, Loveland P, Kanellakis P, Cao A, Kallies A, Huang AL, Peter K, Toh BH, Bobik A (2021) Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur Heart J 42(9):938–947. https://doi.org/10.1093/eurheartj/ehaa995
Dutta P, Courties G, Wei Y et al (2012) Myocardial infarction accelerates atherosclerosis. Nature 487(7407):325–329. https://doi.org/10.1038/nature11260
Yaku H, Fudim M, Shah SJ (2024) Role of splanchnic circulation in the pathogenesis of heart failure: State-of-the-art review. J Cardiol 83(5):330–337. https://doi.org/10.1016/j.jjcc.2024.02.004
Fudim M, Zirakashvili T, Shaburishvili N, Shaishmelashvili G, Sievert H, Sievert K, Reddy VY, Engelman ZJ, Burkhoff D, Shaburishvili T, Shah SJ (2022) Transvenous right greater splanchnic nerve ablation in heart failure and preserved ejection fraction: First-in-human study. JACC Heart Fail 10(10):744–752. https://doi.org/10.1016/j.jchf.2022.05.009
Málek F, Gajewski P, Zymliński R, Janczak D, Chabowski M, Fudim M, Martinca T, Neužil P, Biegus J, Mates M, Krüger A, Skalský I, Bapna A, Engelman ZJ, Ponikowski PP (2021) Surgical ablation of the right greater splanchnic nerve for the treatment of heart failure with preserved ejection fraction: First-in-human clinical trial. Eur J Heart Fail 23(7):1134–1143. https://doi.org/10.1002/ejhf.2209
Di Sabatino A, Carsetti R, Corazza GR (2011) Post-splenectomy and hyposplenic states. Lancet 378(9785):86–97. https://doi.org/10.1016/s0140-6736(10)61493-6
Luoto TT, Pakarinen MP, Koivusalo A (2016) Long-term outcomes after pediatric splenectomy. Surgery 159(6):1583–1590. https://doi.org/10.1016/j.surg.2015.12.014
Li Y, Stone JR (2016) The impact of splenectomy on human coronary artery atherosclerosis and vascular macrophage distribution. Cardiovasc Pathol 25(6):453–460. https://doi.org/10.1016/j.carpath.2016.08.001
Aviram M, Brook JG, Tatarsky I, Levy Y, Carter A (1986) Increased low-density lipoprotein levels after splenectomy: A role for the spleen in cholesterol metabolism in myeloproliferative disorders. Am J Med Sci 291(1):25–28. https://doi.org/10.1097/00000441-198601000-00006
Schwabl P, Seeland BA, Riedl F, Schubert TL, Konigshofer P, Brusilovskaya K, Petrenko O, Hofer B, Schiefer AI, Trauner M, Peck-Radosavljevic M, Reiberger T (2022) Splenectomy ameliorates portal pressure and anemia in animal models of cirrhotic and non-cirrhotic portal hypertension. Adv Med Sci 67(1):154–162. https://doi.org/10.1016/j.advms.2022.02.005
Borsani O, Asano T, Boisson B, Fraticelli S, Braschi-Amirfarzan M, Pietra D, Casetti IC, Vanni D, Trotti C, Borghesi A, Casanova JL, Arcaini L, Rumi E (2022) Isolated congenital asplenia: An overlooked cause of thrombocytosis. Am J Hematol 97(8):1110–1115. https://doi.org/10.1002/ajh.26522
Kristinsson SY, Gridley G, Hoover RN, Check D, Landgren O (2014) Long-term risks after splenectomy among 8,149 cancer-free American veterans: A cohort study with up to 27 years follow-up. Haematologica 99(2):392–398. https://doi.org/10.3324/haematol.2013.092460
Crary SE, Buchanan GR (2009) Vascular complications after splenectomy for hematologic disorders. Blood 114(14):2861–2868. https://doi.org/10.1182/blood-2009-04-210112
Tang Y, Lu W, Zhang Z, Zuo P, Ma G (2015) Hypersplenism: An independent risk factor for myocardial remodeling in chronic heart failure patients. Int J Clin Exp Med 8(4):5197–5206. https://www.ncbi.nlm.nih.gov/pubmed/26131093
Anand IS, Gupta P (2018) Anemia and iron deficiency in heart failure: Current concepts and emerging therapies. Circulation 138(1):80–98. https://doi.org/10.1161/CIRCULATIONAHA.118.030099
Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P (2013) Iron deficiency and heart failure: Diagnostic dilemmas and therapeutic perspectives. Eur Heart J 34(11):816–829. https://doi.org/10.1093/eurheartj/ehs224
Gertler C, Jauert N, Freyhardt P, Valentova M, Aland SC, Walter-Rittel TC, Unterberg-Buchwald C, Placzek M, Ding-Reinelt V, Bekfani T, Doehner W, Hasenfuss G, Hamm B, Sandek A (2023) Magnetic resonance imaging of organ iron before and after correction of iron deficiency in patients with heart failure. ESC Heart Fail 10(3):1847–1859. https://doi.org/10.1002/ehf2.14329
Nagelkerke SQ, Bruggeman CW, den Haan JMM, Mul EPJ, van den Berg TK, van Bruggen R, Kuijpers TW (2018) Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-gamma receptors. Blood Adv 2(8):941–953. https://doi.org/10.1182/bloodadvances.2017015008
Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, Pluhacek T, Spatenka J, Kovalcikova J, Drahota Z, Kautzner J, Pirk J, Houstek J (2017) Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur J Heart Fail 19(4):522–530. https://doi.org/10.1002/ejhf.640
Nunez J, Minana G, Cardells I et al (2020) Noninvasive imaging estimation of myocardial iron repletion following administration of intravenous iron: The myocardial-IRON trial. J Am Heart Assoc 9(4):e014254. https://doi.org/10.1161/JAHA.119.014254
Klip IT, Jankowska EA, Enjuanes C, Voors AA, Banasiak W, Bruguera J, Rozentryt P, Polonski L, van Veldhuisen DJ, Ponikowski P, Comin-Colet J, van der Meer P (2014) The additive burden of iron deficiency in the cardiorenal-anaemia axis: Scope of a problem and its consequences. Eur J Heart Fail 16(6):655–662. https://doi.org/10.1002/ejhf.84
Yamauchi T, Sakata Y, Takada T, Nochioka K, Miura M, Tadaki S, Ushigome R, Sato K, Onose T, Tsuji K, Abe R, Takahashi J, Miyata S, Shimokawa H, investigators C- (2015) Prognostic impact of anemia in patients with chronic heart failure- with special reference to clinical background: Report from the CHART-2 study. Circ J 79(9):1984–1993. https://doi.org/10.1253/circj.CJ-15-0174
Savarese G, Jonsson A, Hallberg AC, Dahlstrom U, Edner M, Lund LH (2020) Prevalence of, associations with, and prognostic role of anemia in heart failure across the ejection fraction spectrum. Int J Cardiol 298:59–65. https://doi.org/10.1016/j.ijcard.2019.08.049
Silverberg D, Wexler D, Blum M, Wollman Y, Iaina A (2003) The cardio-renal anaemia syndrome: Does it exist? Nephrol Dial Transplant 18(Suppl 8):viii7-12. https://doi.org/10.1093/ndt/gfg1084
Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI (2020) Iron deficiency in chronic kidney disease: Updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol 31(3):456–468. https://doi.org/10.1681/ASN.2019020213
Haase VH (2013) Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev 27(1):41–53. https://doi.org/10.1016/j.blre.2012.12.003
Haase VH (2011) (2021) Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int Suppl 11(1):8–25. https://doi.org/10.1016/j.kisu.2020.12.002
Nakamura M, Imamura T, Sobajima M, Kinugawa K (2023) Initial experience of hypoxia-inducible factor prolyl hydroxylase inhibitors in patients with heart failure and renal anemia. Heart Vessels 38(2):284–290. https://doi.org/10.1007/s00380-022-02181-1
Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, Luscher TF, Bart B, Banasiak W, Niegowska J, Kirwan BA, Mori C, von Eisenhart RB, Pocock SJ, Poole-Wilson PA, Ponikowski P, Investigators F-HT (2009) Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 361(25):2436–2448. https://doi.org/10.1056/NEJMoa0908355
Anker SD, Kirwan BA, van Veldhuisen DJ, Filippatos G, Comin-Colet J, Ruschitzka F, Luscher TF, Arutyunov GP, Motro M, Mori C, Roubert B, Pocock SJ, Ponikowski P (2018) Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: An individual patient data meta-analysis. Eur J Heart Fail 20(1):125–133. https://doi.org/10.1002/ejhf.823
Ponikowski P, Kirwan BA, Anker SD et al (2020) Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: A multicentre, double-blind, randomised, controlled trial. Lancet 396(10266):1895–1904. https://doi.org/10.1016/S0140-6736(20)32339-4
Bolger AP, Bartlett FR, Penston HS, O’Leary J, Pollock N, Kaprielian R, Chapman CM (2006) Intravenous iron alone for the treatment of anemia in patients with chronic heart failure. J Am Coll Cardiol 48(6):1225–1227. https://doi.org/10.1016/j.jacc.2006.07.015
Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, Teoh H, Leiter LA, Zinman B, Juni P, Zuo F, Mistry N, Thorpe KE, Goldenberg RM, Yan AT, Connelly KA, Verma S (2020) Effect of empagliflozin on erythropoietin levels, iron stores, and red blood cell morphology in patients with type 2 diabetes mellitus and coronary artery disease. Circulation 141(8):704–707. https://doi.org/10.1161/CIRCULATIONAHA.119.044235
Docherty KF, Welsh P, Verma S et al (2022) Iron deficiency in heart failure and effect of dapagliflozin: Findings from DAPA-HF. Circulation 146(13):980–994. https://doi.org/10.1161/CIRCULATIONAHA.122.060511
Kommu S (2024) The role of SGLT2 inhibitors on heart failure outcomes in nondiabetic patients: A systematic review and meta-analysis of randomized controlled trials. J Cardiovasc Pharmacol 83(2):158–166. https://doi.org/10.1097/fjc.0000000000001511
Packer M (2023) Potential interactions when prescribing SGLT2 inhibitors and intravenous iron in combination in heart failure. JACC Heart Fail 11(1):106–114. https://doi.org/10.1016/j.jchf.2022.10.004
Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA (2020) Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res 126(9):1260–1280. https://doi.org/10.1161/CIRCRESAHA.120.315937
Wu J, Dong E, Zhang Y, Xiao H (2021) The role of the inflammasome in heart failure. Front Physiol 12:709703. https://doi.org/10.3389/fphys.2021.709703
Chen GY, Nunez G (2010) Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol 10(12):826–837. https://doi.org/10.1038/nri2873
Nakayama H, Otsu K (2013) Translation of hemodynamic stress to sterile inflammation in the heart. Trends Endocrinol Metab 24(11):546–553. https://doi.org/10.1016/j.tem.2013.06.004
Li Y, Huang X, Wang J, Huang R, Wan D (2020) Regulation of iron homeostasis and related diseases. Mediators Inflamm 2020:6062094. https://doi.org/10.1155/2020/6062094
Leuschner F, Rauch PJ, Ueno T et al (2012) Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209(1):123–137. https://doi.org/10.1084/jem.20111009
Voulgari PV, Kolios G, Papadopoulos GK, Katsaraki A, Seferiadis K, Drosos AA (1999) Role of cytokines in the pathogenesis of anemia of chronic disease in rheumatoid arthritis. Clin Immunol 92(2):153–160. https://doi.org/10.1006/clim.1999.4736
Isidoro CA, Deniset JF (2023) The role of macrophage subsets in and around the heart in modulating cardiac homeostasis and pathophysiology. Front Immunol 14:1111819. https://doi.org/10.3389/fimmu.2023.1111819
Halade GV, Norris PC, Kain V, Serhan CN, Ingle KA (2018) Splenic leukocytes define the resolution of inflammation in heart failure. Sci Signal 11(520):eaao1818. https://doi.org/10.1126/scisignal.aao1818
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510(7503):92–101. https://doi.org/10.1038/nature13479
Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, Rudd JH, Fayad ZA, Lehrer-Graiwer J, Korsgren M, Figueroa AL, Fredrickson J, Rubin B, Hoffmann U, Truong QA, Min JK, Baruch A, Nasir K, Nahrendorf M, Tawakol A (2015) Splenic metabolic activity predicts risk of future cardiovascular events: Demonstration of a cardiosplenic axis in humans. JACC Cardiovasc Imaging 8(2):121–130. https://doi.org/10.1016/j.jcmg.2014.10.009
Abboud A, Nguonly A, Bean A, Brown KJ, Chen RF, Dudzinski D, Fiseha N, Joice M, Kimaiyo D, Martin M, Taylor C, Wei K, Welch M, Zlotoff DA, Januzzi JL, Gaggin HK (2021) Rationale and design of the preserved versus reduced ejection fraction biomarker registry and precision medicine database for ambulatory patients with heart failure (PREFER-HF) study. Open Heart 8(2). https://doi.org/10.1136/openhrt-2021-001704
McKim DB, Yin W, Wang Y, Cole SW, Godbout JP, Sheridan JF (2018) Social stress mobilizes hematopoietic stem cells to establish persistent splenic myelopoiesis. Cell Rep 25(9):2552-2562 e2553. https://doi.org/10.1016/j.celrep.2018.10.102
Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA (2012) An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 7(5):e36814. https://doi.org/10.1371/journal.pone.0036814
Schiattarella GG, Alcaide P, Condorelli G, Gillette TG, Heymans S, Jones EAV, Kallikourdis M, Lichtman A, Marelli-Berg F, Shah S, Thorp EB, Hill JA (2022) Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nat Cardiovasc Res 1(3):211–222. https://doi.org/10.1038/s44161-022-00032-w
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116(7):2672–2680. https://doi.org/10.1073/pnas.1821022116
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Doring Y, Drechsler M, Soehnlein O, Weber C (2015) Neutrophils in atherosclerosis: From mice to man. Arterioscler Thromb Vasc Biol 35(2):288–295. https://doi.org/10.1161/ATVBAHA.114.303564
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S (2017) Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38(3):187–197. https://doi.org/10.1093/eurheartj/ehw002
Fisher SM, Murally AR, Rajabally Z, Almas T, Azhar M, Cheema FH, Malone A, Hasan B, Aslam N, Saidi J, O’Neill J, Hameed A (2024) Large animal models to study effectiveness of therapy devices in the treatment of heart failure with preserved ejection fraction (HFpEF). Heart Fail Rev 29(1):257–276. https://doi.org/10.1007/s10741-023-10371-w
Li H, Xia YY, Xia CL, Li Z, Shi Y, Li XB, Zhang JX (2022) Mimicking metabolic disturbance in establishing animal models of heart failure with preserved ejection fraction. Front Physiol 13:879214. https://doi.org/10.3389/fphys.2022.879214
Ranjbarvaziri S, Zeng A, Wu I, Greer-Short A, Farshidfar F, Budan A, Xu E, Shenwai R, Kozubov M, Li C, Van Pell M, Grafton F, MacKay CE, Song X, Priest JR, Argast G, Mandegar MA, Hoey T, Yang J (2024) Targeting HDAC6 to treat heart failure with preserved ejection fraction in mice. Nat Commun 15(1):1352. https://doi.org/10.1038/s41467-024-45440-7
ClinicalTrials.gov (2022) Empagliflozin outcome trial in patients with chronic heart failure with preserved ejection fraction (EMPEROR-Preserved). Retrieved May 9 from https://clinicaltrials.gov/study/NCT03057951
Abraham WT, Lindenfeld J, Ponikowski P et al (2021) Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur Heart J 42(6):700–710. https://doi.org/10.1093/eurheartj/ehaa943
Anker SD, Butler J, Filippatos G et al (2021) Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med 385(16):1451–1461. https://doi.org/10.1056/NEJMoa2107038
Prabhu SD (2018) The cardiosplenic axis is essential for the pathogenesis of ischemic heart failure. Trans Am Clin Climatol Assoc 129:202–214. https://www.ncbi.nlm.nih.gov/pubmed/30166715
Zhu J, Chen S, Wang J, Zhang C, Zhang W, Liu P, Ma R, Chen Y, Yao Z (2013) Splenectomy increases the survival time of heart allograft via developing immune tolerance. J Cardiothorac Surg 8:129. https://doi.org/10.1186/1749-8090-8-129
Li W, Li L, Li W, Chopp M, Venkat P, Zacharek A, Chen Z, Landschoot-Ward J, Chen J (2020) Spleen associated immune-response mediates brain-heart interaction after intracerebral hemorrhage. Exp Neurol 327:113209. https://doi.org/10.1016/j.expneurol.2020.113209
O’Neill HC, Lim HK (2023) Skeletal stem/progenitor cells provide the niche for extramedullary hematopoiesis in spleen. Front Physiol 14:1148414. https://doi.org/10.3389/fphys.2023.1148414
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD (2014) Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ Res 114(2):266–282. https://doi.org/10.1161/CIRCRESAHA.113.301720
Tian Y, Miao B, Charles EJ, Wu D, Kron IL, French BA, Yang Z (2018) Stimulation of the Beta2 adrenergic receptor at reperfusion limits myocardial reperfusion injury via an interleukin-10-dependent anti-inflammatory pathway in the spleen. Circ J 82(11):2829–2836. https://doi.org/10.1253/circj.CJ-18-0061
Jaiswal S, Fontanillas P, Flannick J, Ebert Bl et al (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371(26):2488–2498. https://doi.org/10.1056/NEJMoa1408617
Hulsmans M, Sager HB, Roh JD, Nahrendorf M et al (2018) Cardiac macrophages promote diastolic dysfunction. J Exp Med 215(2):423–440. https://doi.org/10.1084/jem.20171274
Acknowledgements
The authors would like to thank Ryota Morimoto, MD, PhD; Akinori Sawamura, MD, PhD; Toru Kondo, MD, PhD; Shingo Kazama, MD, PhD; Ryota Ito, MD; and Shin Nagai, MD, for their useful discussion and/or helpful comments on the manuscript.
Funding
Open Access funding provided by Nagoya University. This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS KAKENHI) (grant number JP21K16085 to H.H.).
Author information
Authors and Affiliations
Contributions
Conceptualization, formal analysis, funding acquisition, methodology, project administration, supervision, visualization, and writing—original draft preparation: Hiroaki Hiraiwa; data curation, investigation and resources: Hiroaki Hiraiwa and Yoshimitsu Yura; writing—review and editing: Hiroaki Hiraiwa, Yoshimitsu Yura, Takahiro Okumura, and Toyoaki Murohara; all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Hiroaki Hiraiwa declares the following: research grants from Konica Minolta, Inc., Konica Minolta Science and Technology Foundation, and Kowa Life Science Foundation. Yoshimitsu Yura declares the following: research grants from Japan Foundation for Applied Enzymology, Kowa Life Science Foundation, MSD Life Science Foundation, Mitsubishi Foundation, Suzuken Memorial Foundation, the Hori Science and Arts Foundation, Mochida Foundation, Takeda Science Foundation, Sakakibara Heart Foundation, and Japan Heart Foundation. Takahiro Okumura declares the following: research grants from Ono Pharmaceutical Co. Ltd., Amgen Astellas BioPharma K.K., Pfizer Japan Inc., Alnylam Pharmaceuticals, Inc., and Alexion Pharmaceuticals, Inc. Takahiro Okumura received lecture fees from Ono Pharmaceutical Co. Ltd., Otsuka Pharma Ltd., Novartis Pharma K.K., and AstraZeneca K.K. Toyoaki Murohara declares the following: an unrestricted research grant from the Department of Cardiology, Nagoya University Graduate School of Medicine, from Astellas Pharma Inc., Daiichi-Sankyo Co. Ltd., Dainippon Sumitomo Pharma Co. Ltd., Kowa Co. Ltd., MSD K.K., Mitsubishi Tanabe Pharma Co., Nippon Boehringer Ingelheim Co. Ltd., Novartis Pharma K.K., Otsuka Pharma Ltd., Pfizer Japan Inc., Sanofi-Aventis K.K., Takeda Pharmaceutical Co. Ltd., and Teijin Pharma Ltd. All authors declare that they have no relationships with industry relevant to the contents of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hiraiwa, H., Yura, Y., Okumura, T. et al. Interplay of the heart, spleen, and bone marrow in heart failure: the role of splenic extramedullary hematopoiesis. Heart Fail Rev 29, 1049–1063 (2024). https://doi.org/10.1007/s10741-024-10418-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-024-10418-6