
Abstract
The aim of this study was to reveal the intra and interspecies differences between Triticum baeoticum and Triticum urartu using Diversity Arrays Technology sequencing (DArTseq) on 94 accessions representing Turkish populations. Seeds were gathered from the US Department of Agriculture, and from the Turkish Seed Gene Bank. Isolated and purified DNA samples were sent to Diversity Arrays Technologies for DArTseq. After the necessary quality filtering, a total of 16,898 and 100,103 loci were obtained respectively from the single nucleotide polymorphism (SNP) and SilicoDArT datasets. ADMIXTURE software was used to reveal the intra and interspecies population structures. Analysis of molecular variance was carried out to reveal the variance between the populations of the T. urartu and T. baeoticum species. Principal coordinate analysis was conducted to visualize the main sources of variation between the populations on a 2-dimensional plane. To reveal the evolutionary relationship, SNP dataset was used to reconstruct the phylogenetic dendrograms by using the maximum likelihood statistical method and the unweighted pair group method with arithmetic mean clustering algorithm. As a result of this study, the accessions of T. urartu and T. baeoticum species formed separate clusters and revealed as two different species. In line with the results obtained, it is obvious that the identification of some accessions should be re-evaluated. The results demonstrated that DArTseq, is a fast, low-cost, and high-accuracy method that can be used in species and population discrimination and an effective tool for Gene Bank management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
About 12,000 years ago, agricultural practices appeared in the fertile crescent region and then spread to world (Kilian et al. 2010). The center of wheat diversity and initial domestication lies in the Fertile Crescent (Zeibig et al. 2022). Türkiye’s southeastern Anatolia region is within this fertile crescent, and Triticum baeoticum Boiss. T. dicoccoides (Korn ex Aschers. et Graebn.)Thell, T. urartu Thum. ex Gandil. and T. araraticum Jakubz. species grow naturally in this region. Diploid wheats (AA) belong to the oldest cultivated plant species. Wild representatives of the diploid wheat group are Triticum baeoticum and Triticum urartu (Zeibig et al. 2022). Triticum monococcum was among the first crops domesticated in the Fertile Crescent starting from the wild progenitor T. baeoticum (Kilian et al. 2010). Triticum urartu is the ancestral progenitor of the A genome of T. aestivum (BBAADD) (Ling et al. 2018). According to Wang et al. (2022) T. urartu originated from northwestern Syria and genetic changes were revealed between the eastern Mediterranean coastal and Mesopotamian-Transcaucasian populations.
The genus Triticum belongs to the tribe Triticeae Dumort, which includes 18 genera and 80 species. Triticeae is one of the most important tribes of the family Poaceae, which includes economically important grains besides wheat, such as barley (Hordeum vulgare L.) and rye (Secale cereale L.) (Edet et al. 2018). The genus Triticum, which is closely related to the genus Aegilops L., has been considered a separate genus by most researchers since Linneaus (1753) (Goncharov 2011). There are 10 species of the genus Triticum in Türkiye (Tan 1985): T. urartu, T. baeoticum, T. monococcum, T. dicoccon, T. dicoccoides, T. araraticum, T. turgidum L., T. carthlicum Nevski, T. durum, and T. aestivum. Among these species, T. baeoticum, T. urartu, T. araticum, and T. dicoccoides are wild growing species in flora of Türkiye. Triticum baeoticum has a wide distribution range in Türkiye, while T. urartu is naturally distributed only in southeastern Anatolia (Fig. 1). We have followed the classification purposed by Dorofeev et al. (1979) in our research. Triticum urartu and T. baeoticum species are morphologically very close and it is difficult to identify these species correctly using morphological characters. The extensive revision of the genus Triticum was performed by Cabi (2010). When the descriptions of T. urartu and T. baeoticum given in Cabi (2010) were examined, the spike height is 5–14 cm in T. baeoticum and 6–12 cm in T. urartu; spikelets are 12–17 and 12–16 mm, respectively; both are rectangular and have 2–3 flowers; glumes are (4–)6–10 and 8–11 mm, respectively; lemmas are 10–14 and 10–13 mm, respectively; lemma awns are up to 8 and 7 cm, respectively; and the anther length is ca. 3–5 and 2–4 mm, respectively. As can be seen, the characteristics of the characters in the existing genre descriptions are intertwined with each other. Therefore, it is difficult to make precise identifications and a deeper characterization is essential for seedbanks in order to prevent misidentification.

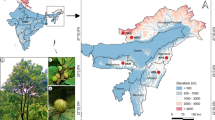
Distribution map of the accessions used in the study
In addition to morphological characterizations, molecular approaches have been applied to reveal the taxonomy of these two species. Golovnina et al. (2007) and Goncharov et al. (2009) examined the phylogenetic relationships of cultivated and natural species of the genus Triticum, and the results showed that the diploids, tetraploids, and hexaploids were clustered separately. Dizkirici et al. (2016) revealed that the tree resulting from the sequence analysis of the matK region, T. baeoticum, T. urartu, and T. monococcum were clustered together. In the phylogenetic tree reconstructed with matK and internal transcribed spacer (ITS) regions, T. baeoticum, T. urartu, and T. monococcum were clustered together as well (Dizkirici et al. 2016). According to the phylogeny tree based on the ITS2 intronic DNA region Triticum urartu and T. baeoticum were clustered together in Safari and Mehrabi’s research (2019). All mentioned Sanger sequencing-based researches show that T. urartu and T. baeoticum are closely related species.
Today, intra and interspecies relationships can be examined in more detail by scanning similarities/differences in a much larger amount of DNA sequences, rather than analysis based on a limited exonic and intronic genetic markers. Genotyping by sequencing (GBS), one of the leading approaches, is applied by sequencing many target regions throughout the genome with one or more cutting enzymes (Wallace and Mitchel 2017). According to Hyun et al. (2020), GBS-derived single nucleotide polymorphisms (SNPs) could be used efficiently in gene bank management (GBM) to classify Triticum species and subspecies that are very difficult to distinguish by their morphological characters. Diversity Arrays Technology sequencing (DArTseq) analysis, one of the genotyping methods via sequencing, can also be applied to wild species as reference genome information is not required (Hyun et al. 2020). In addition, since DArTseq performs sequencing with greater read depth at the whole genome level, it creates markers with less missing data and high reproducibility compared to other GBS approaches (Allan et al. 2020). Edet et al. (2018) investigated the phylogeny of Aegilops and Triticum species in which they examined the genomic relationships of the tribe Triticeae. Among the materials in this study, T. urartu and T. baeoticum were represented by one accession for each species (Edet et al. 2018). Wang et al. (2022) investigated the population genomics of T. urartu via the genome-wide sequencing of 59 representative accessions collected around the world. In their research, 10 accessions from Türkiye were gathered from the USDA and 4 of these accessions were in common with the current study (Wang et al. 2022). Abbasov et al. (2020) used DArTseq technology and SNP markers to evaluate the genetic diversity among 150 accessions of different Aegilops species from Azerbaijan. They reported a sufficient level of genetic diversity of Aegilops from different ecogeographical regions of the country (Abbasov et al. 2020). Baloch et al. (2017) performed whole genome DArTseq and SNP analysis to perform a genetic diversity assessment in durum wheat from central fertile crescent. Brandolini et al. (2016) conducted a research with DArTseq markers on 136 landraces of einkorn (T. monococcum) accessions from various countries in Europe, 9 of which were wild einkorn and 3 were T. urartu, to determine the geographical differentiation of einkorn wheat and possible migration routes. They confirmed that T. monococcum separated from the Karacadağ population of T. urartu, meaning that einkorn was cultured in the Karacadağ region. The study material comprised 2 accessions from Türkiye (Brandolini et al. 2016). Fufa et al. (2023) performed genetic diversity and population structure analyses in tetraploid wheat (T. turgidum spp.) germplasm from Ethiopia based on DArTSeq markers. They found that high genetic diversity was detected in the Ethiopian tetraploid wheat germplasm, which could be utilized for future wheat breeding programs (Fufa et al. 2023). Sansaloni et al. (2020) investigated approximately 80,000 wheat accessions from the wheat collections of the International Maize and Wheat Improvement Center and International Center for Agricultural Research in the Dry Areas with DarTseq-based SNP and Silico-DArT markers and revealed the genetic diversity of the wheats. They included 3903 accessions from wild relative species of genera Triticum and Aegilops. However, in their study, only 2 T. urartu accessions originating from Türkiye were included among the wild related species (Sansaloni et al. 2020).
DArTseq and SNP analysis are efficient tools for genetic characterization and GBM. Southeastern Anatolia is considered as the site of einkorn wheat domestication (Haas et al. 2019). Wild populations from Türkiye play a key role in understanding the genetic diversity of T. urartu and T. baeoticum. The accessions preserved in the Turkish Seed Gene Bank (TSGB) have not been used as material in any of the existing research. In the current research, the population structure and phylogenetic relationships were used in order to better understand the accessions and improve the germplasm management with DArTseq and SNP analysis. With this study, detailed genetic characterization of Turkish T. urartu and T. baeoticum accessions were given and the possible misidentifications among the germplasm stored in the USDA and TSGB were revealed.
Materials and methods
Plant material
A total of 94 accessions, comprising 59 T. urartu (hereafter TU) and 35 T. baeoticum (hereafter TB), were used as the study materials (Supplementary file). TU accessions were obtained from the US Department of Agriculture (USDA) and accessions of TB were obtained from the TSGB. All of the accessions were of Turkish origin.
DNA extraction and DArT genotyping
Seeds were germinated in the TSGB’s climate chambers and 100 mg of fresh leaf tissue was taken from seedlings and used for genomic DNA isolation, which was performed according to the protocol provided by Diversity Arrays Technology Pty Ltd. (DArT Pty Ltd.) (Canberra, Australian Capital Territory, Australia; https://ordering.diversityarrays.com/files/DArT_DNA_isolation.pdf). Leaf tissue was taken from one plant to represent each accession. The quality and quantity of the extracted DNA were analyzed using Nanodrop Lite (Thermo Fisher Scientific Inc., Waltham, MA, USA) and confirmed on 0.8% agarose gel. DNA samples were normalized according to the original sampling standards of DArT Pty Ltd. and DNA concentrations of 50 ng and then sent to DArT Pty Ltd. (http://www.diversityarrays.com/) for DArTseq. All of the images from the DArTseq and SNP platforms were analyzed using DArTsoft v.7.4.7 (DArT Pty Ltd.). The SilicoDArT and SNP markers were scored using DArTsoft as binary data (1/0), indicating the presence or absence of a marker in the genomic representation of each sample. The DArTsoft automatically computed several quality parameters for each SilicoDArT and SNP marker, such as the call rate, polymorphic information content (PIC), and reproducibility of both markers. The SNP and SilicoDArT datasets were obtained after the filtering functions in the dartR package, with a minor allele frequency of 5%, call rate of 85%, and repeatability rate of 95%. In order to improve the quality of the SNP and SilicoDArt datasets in terms of missing data, the missing data were filled in without compromising the integrity of the dataset using the “gl.impute” function in the dartR package. The observed heterozygosity (Ho), expected heterozygosity (Hs), total heterozygosity (Ht), Dst (Ht–Hs), Dstp (np/(np–1), and other fixation indices were calculated using the gl.basic.stats function of the dartR package in R programming language.
Population structure and analysis of molecular variance (AMOVA)
As a result of the DArTSeq, there were 56,188 loci in the SNP dataset and 105,260 loci in the SilicoDArT dataset. After filtering the SNP dataset according to the call rate, the minimum allele frequency (MAF) and repeatability coefficients, a total of 16,898 SNPs and 100,103 SilicoDArT markers were obtained. Two distinct methods were applied to assess the population structure: principal coordinate analysis (PCoA) and the Bayesian clustering approach of ADMIXTURE. PCoA was conducted using R v. 4.3.0 with ape package v. 5.7.1 (R Core Team 2023). The default parameters of the find.clusters function were used to determine K, maintaining all principal components (PCs). The s.optim.pca function within this framework was employed to confirm the appropriate number of PCs to retain. Additionally, to delve into the population structure, ADMIXTURE analysis was performed on 94 individuals using the ADMIXTURE tool (Alexander et al. 2009). ADMIXTURE adopts the likelihood model embedded in structure and runs considerably faster (Alexander et al. 2009). The analysis was executed in unsupervised mode with 8 threads using ADMIXTURE-Linux-1.3.0, exploring K values ranging from 1 to 10 to obtain the optimum K value, which was determined by computing the cross-validation error for each K using the 10-fold-cv option. As a result, the cross-validation and K values with the lowest cross-validation error were determined as 5, and the K value with the highest LogLikelihood value was determined as 2.
A detailed AMOVA was conducted on the filtered SNP and SilicoDArT markers using the ‘stampAmova’ function from the ‘StAMPP’ package, with 100 permutations in the R programming language. Through this analysis, the effects of these markers on the genetic diversity and inter or intra-population variations were comprehensively examined. PCoA, which provided coordinate-based visualization of the data on the distances between the principal elements in the SilicoDArT and SNP datasets, was used after calculating the distance matrix between the PCs in the SilicoDArT and SNP datasets using the “vegdist” and “cmdscale” functions in the “vegan” package in R programming language, and then visualized using the “ggplot2” library.
Genetic diversity and phylogeny
Unweighted pair group method with arithmetic mean (UPGMA) and maximum likelihood (ML) analyses were performed to reconstruct the dendrogram and phylogeny. SNPs were aligned using Molecular Evolutionary Genetics Analysis software v.11 (Tamura et al. 2021). In the UPGMA analysis, the p-distance was employed to calculate the distance method, which determines the percentage difference between 2 sequences. The pairwise deletion method was utilized to handle gaps or missing data in each sequence pair, excluding such positions from the calculations. Additionally, to assess the tree’s reliability, 1000 bootstrap values were applied. For the ML phylogeny test, the nucleotide was chosen as the substitution type, and the general time reversible model was used as the model or method. Ratios between the nucleotide positions were modelled with the gamma distribution and 5 separate gamma categories were determined. For missing data and gaps, the Use all Sites method was employed. The nearest-neighbor-interchange (NNI) was chosen as the heuristic method for ML and neighbor-joining method was chosen as the starting tree.
Results
Genotyping and SNP calling
Among a total of 16,898 SNPs and 100,103 SilicoDArT markers after filtering, the call rate increased to 0.961 and the minor allele frequency increased to 0.264 and 1. The Ho (observed heterozygosity) was 0.03 for SNP dataset and 0.29 for the SilicoDArT dataset. According to the basic statistics data obtained from the SNP dataset, the Ho rate in the SNP dataset was 0.03, while the Hs (expected heterozygosity) rate was 0.20, which means that the genetic diversity between TU and TB populations was high (Table 1). The fact that the Dst (genetic difference between populations) and Dstp (genetic difference between corrected populations) data were 0.21 and 0.42, respectively, also supported this conclusion. According to the data obtained from the basic statistics, the investigated accessions belong to 2 major groups: TU and TB.
Table 2 provides a summary of the SNP and SilicoDArT marker distribution and densities of the TU and TB chromosomes. According to the SNP distribution, the highest number of SNPs was revealed in chromosome5A (1776) and the lowest number was in chromosome4B (121). The highest number of SilicoDArT markers was observed in chromosome7A (7942) and lowest number was in chromosome4D (827).
In Table 2 and Fig. 2, the distribution of SNP and SilicoDArT markers across various chromosomes from Chr1A to Chr7D is illustrated. Chromosome Chr1A exhibits a high count with 1,291 SNP markers and 5,050 SilicoDArT markers. In contrast, chromosomes Chr4D and Chr5D contain fewer markers, with 264 and 385 SNP markers respectively. Notably, Chr7A has the highest count of SilicoDArT markers at 7,942. This data highlights the variability in genetic marker distribution, underscoring the diverse genetic profiling that is crucial for chromosomal research in species such as Triticum.

Bar plot of chromosomal distributions of single nucleotide polymorphism (SNP) and Diversity Arrays Technology (SilicoDArT) markers in Triticum urartu (TU) and T. baeoticum (TB) chromosomes
Population structure and AMOVA
Q matrices were visualized according to the K = 2 and K = 5 values determined as a result of the ADMIXTURE analysis performed using the SNP markers. The value K = 2 was chosen because it had the logarithm of the highest ML ratio, and the value K = 5 was chosen because it had the lowest cross-validation error. As a result, the population structure chart with K = 2, which divided the accessions into 2 different populations, can be named as species TU and TB, while the population structure chart with K = 5 made inferences that these 2 different populations could be divided into different subpopulations (Fig. 3).

ADMIXTURE results assuming two and five ancestral populations
The AMOVA of the 94 populations with the SNP and SilicoDArT markers was performed at 2 different hierarchical levels (Table 3). According to the AMOVA results, the sum of square deviations (SSD) and mean square deviations (MSD) among the TU and TB accessions were higher in the SNP markers than in the SilicoDArT markers (27.29 and 5.65, respectively). According to the AMOVA results of the SNPs, the high Fst value among the accessions (0.82) indicated significant genetic differentiation between the TU and TB accessions. The P values suggested that the observed genetic variation among the TU and TB accessions was statistically significant (P = 0.00), but the variation within the TU and TB accessions (error) was not (df = 92.00). The phi-statistics (0.83) and variance coefficients (43.94) provided information about the proportion of genetic variance among the TU and TB accessions. According to the AMOVA results of the SilicoDArT markers, the Fst value among the accessions (0.79) suggested some genetic differentiation between the TU and TB accessions. Similar to the SNP analysis, the P value for the variation among the TU and TB accessions was indicating statistical significance (P = 0.00). The phi-statistics (0.79) and variance coefficients (43.94) provided additional insight into the proportion of genetic variance among the TU and TB accessions. Variance coefficient indicates that both SNP and SilicoDArT data provide a similar level of information for detecting genetic variation. This demonstrates that both data types are effective in reflecting genetic structure within and among populations and are reliable tools for assessing genetic diversity. Analyzing SNP and SilicoDArT data with AMOVA is used to determine whether genetic variation is more prevalent within populations or between them. The similar results from different genotyping platforms, SNP and SilicoDArT, suggest that both can be used for various research purposes. Both the SNP and SilicoDArT analyses showed significant genetic differentiation among the TU and TB accessions. The P values indicated that the observed genetic variation among the TU and TB accessions was statistically significant. Phi-statistics and variance coefficients provide measures of the proportion of genetic variance at different levels.
As a result of both PcoAs performed using the PC1 and PC2 matrices obtained by the SNP and SilicoDArT markers (Fig. 4), the TU and TB accessions were clustered as 2 separate clusters. While TU accessions with code numbers 38 and 63 were clustered with accessions belonging to TB species, TB accessions with code numbers 15, 18, 26, 34 and 35 were clustered with accessions belonging to TU accessions.

Single nucleotide polymorphism (SNP) and Diversity Arrays Technology (SilicoDArT) principal coordinate analysis (PCoA) plots
Phylogenetic relationships
The ML and UPGMA clustering methods were used to examine the phylogenetic relationships of individuals representing the accessions of TU and TB species. In the phylogenetic dendrogram created with the ML statistical method (Fig. 5), group 1 consisted of 1 accession belonging to TB species; group 2 consisted of 31 accessions in total, 2 belonging to TU species and 29 belonging to TB species; and group 3 consisted of 57 accessions belonging to TU species and 5 belonging to TB species, for a total of 62 accessions.

Maximum likelihood dendrogram based on the single nucleotide polymorphism data
In the dendrogram created with the UPGMA clustering algorithm (Fig. 6), the TU and TB species were divided into 2 main groups. Group 1 consisted of 29 accessions belonging to TB species and 2 belonging to TU species, for a total of 31 accessions. Group 2 consisted of 57 accessions belonging to TU species and 6 belonging to TB species, for a total of 63 accessions.

Unweighted pair group method with arithmetic mean (UPGMA) dendrogram based on the single nucleotide polymorphism (SNP) data
The 94 accessions were divided into 3 different groups in the phylogenetic tree created with the ML statistical method, and 2 groups in the dendrogram created with the UPGMA clustering algorithm. In the tree created according to the ML method, accessions 38 and 63 were grouped together with TB species, while accessions 15, 18, 26, 34 and 35 were grouped in the same group with the TU species.
In the dendrogram created with the UPGMA clustering algorithm, the 94 accessions were divided into 2 groups. Similar to the phylogenetic tree created by the ML statistical method, accessions 38 and 63 were grouped with TB species, while accessions 15, 16, 18, 26, 34 and 35 were grouped with TU species.
Discussion
Cultivated diploid einkorn, T. monococcum ssp. monococcum, was derived from TB (Syn. T. monococcum ssp. boeoticum), a close relative of TU. Although TU itself was never domesticated, it is considered to be the progenitor of the A genome of the hexaploidy wheat, T. aestivum (Haas et al. 2019). The comparative analysis of two pivotal investigations in Triticum species research reveals both complementary and distinctive contributions to the field of agricultural genetics. The first study, focused on establishing a comprehensive phylogeny for the Triticum genus through Genotyping-by-Sequencing (GBS), generated 14,188 high-quality single nucleotide polymorphisms (SNPs) across 283 Triticum-related accessions (Hyun et al. 2020). This endeavor illuminated the complex genetic relationships within the genus, successfully distinguishing 17 species/subspecies and offering an unprecedented resolution of Triticum's evolutionary pathways (Hyun et al. 2020). The objective of Hyun et al. (2020) was not only to map out the phylogenetic tree but also to lay a foundation for future breeding programs by providing a robust genetic framework. In the present study, we have narrowed Hyun et al.’s (2020) scope and investigated the genetic diversity and population structure within Turkish populations of TU and TB. In the exploration of the genetic compability and crossbreeding potential between TU and TB, Fricano et al. (2014) present compelling evidence on the feasibility of developing interspecific introgression lines between these two diploid wheat species. This study underscores the evolutionary significance and the practical implications of such genetic inventions in modern agriculture. Utilizing Diversity Arrays Technology sequencing (DArTSeq) on 94 accessions, it uncovered 16,898 SNPs and 100,103 SilicoDArT markers, marking a significant step towards understanding the intra- and interspecies genetic variations. This research emphasized the practical implications of such genetic insights, particularly for gene bank management and species identification, highlighting the potential for addressing misidentifications within gene banks through high-resolution genomic data. The methodologies of both investigationss, though centered around advanced genotyping technologies, catered to their specific objectives one aiming for a broad genus-wide phylogenetic analysis and the other focusing on detailed population genetics within a subset of species. The findings from these investigations collectively enrich our understanding of Triticum’s genetic diversity and evolutionary history. The first article provides a macro view of the genus's phylogeny, essential for broad-scale genetic research and breeding program optimizations (Hyun et al. 2020). In contrast, our research offers a micro view, delving into the genetic intricacies of specific species within a localized context, thereby facilitating precise conservation and utilization of genetic resources. Together, these investogations underscore the diverse applications of genomic technologies in plant genetics, from enhancing our evolutionary understanding of a major crop genus to refining gene bank collections for better conservation and use of genetic diversity. The crop wild species provide an important source for breeding. The efficient introgression of novel genes from wild relatives to cultivar genotypes greatly depends on the accurate identification of wild species. Despite having standard procedures in gene banks such as the GBM system for the efficient management of plant genetic resources, incorrect classification is not uncommon (Hyun et al. 2020). TU (AuAu) and TB (AbAb) are morphologically very close species, even though they can be genetically easily distinguished. Sanger sequencing methods provide a phylogeny inference; however, they are not efficient tools for discrimination populations (Golovnina et al 2007; Goncharov et al. 2009; Dizkirici et al. 2016; Safari and Mehrabi 2019). The results of Hyun et al. (2020) and our results demonstrated that GBS-derived SNPs could be used efficiently in GBM to classify Triticum species and subspecies that are very difficult to distinguish using their morphological characters.
Using DArTseq and SNP markers is a very common tool for the genetic characterization of crops and their wild relatives. Abbasov et al. (2020) used a total of 61,574 SilicoDArTseq and 30,433 SNP markers when analyzing the genetic diversity in populations of Aegilops species from Azerbaijan. In their research Shannon’s Genetic Diversity Index was used for measuring genetic diversity, Rogers genetic distance was used in cluster analysis and STRUCTURE software was used to reveal the relationships among the Aegilops species (Abbasov et al. 2020). Baloch et al. (2017) obtained a total of 39,568 DArTseq and 20,661 SNP markers to characterize the genetic characteristics of 91 durum wheat land races. The genetic diversity within wheat species, central to the agricultural evolution in the Fertile Crescent and southeastern Anatolia, unfolds through groundbreaking research utilizing advanced genotyping technologies. The investigations on durum wheat landraces from the Central Fertile Crescent and the progenitor species TU and TB, rooted in these historically rich agricultural hubs, illuminate the genetic richness and complexity inherent in these vital crops. By harnessing Diversity Arrays Technology Sequences (DArTseq) and Single Nucleotide Polymorphism (SNP) markers, the investigation into 91 durum wheat landraces unearthed a treasure trove of genetic diversity that defies geographical and traditional expectations. This discovery underscores the notion that agricultural practices, seed exchange, and selection by farmers over millennia have sculpted a genetic panorama that transcends modern national boundaries and environmental barriers. The analysis, employing a vast array of 39,568 DArTseq and 20,661 SNP markers, provided a nuanced understanding of the genetic interplay at work, revealing an unexpected lack of geographical clustering among the durum wheat cultivars. This outcome points to a rich history of gene flow and genetic mixing, likely facilitated by ancient trade routes and the exchange of seeds among diverse communities, further blurring the lines between distinct geographical origins. Techniques such as Neighbor Joining (NJ) and Principal Coordinate Analysis (PCoA) were instrumental in highlighting the shared genetic heritage among the landraces of Türkiye and Syria, suggesting a complex narrative of agricultural development that is intricately linked to human history in the region (Baloch et al. 2017).
Our study focusing on TU and TB, employing DArTSeq to analyze 94 accessions, shed light on the significant genetic differentiation and diversity within these progenitor species. This research not only reinforced the genetic distinction between the species but also unveiled the limitations of morphological classification, as some accessions appeared to have been misclassified under traditional assessments. The use of 16,898 SNPs and 100,103 SilicoDArT markers facilitated a deep dive into the genetic structure, employing methods like ADMIXTURE software for population structure analysis, and analysis of molecular variance (AMOVA), which painted a vivid picture of the genetic landscapes of TU and TB. In our phylogenetic analysis of TB and TU, employing both Maximum-Likelihood and UPGMA methods, significant regional clusters were observed, particularly in Gaziantep and Şanlıurfa. The Maximum-Likelihood method highlighted a notable concentration of TB accessions in groups 2 and 3, with Şanlıurfa displaying a substantial predominance in group 3 with 36 samples. Similarly, the UPGMA analysis confirmed this pattern, additionally revealing significant sample presence in Gaziantep for group 1 with 7 samples, aligning with the findings from Şanlıurfa. These distribution patterns suggest that ecological or geographical factors may be influencing the prevalence and grouping of these species within the regions. These findings are critical for refining the genetic conservation strategies and breeding programs, as they offer a clear demarcation of genetic diversity that is essential for enhancing crop resilience and sustainability. Moreover, the identification of misclassified accessions based on DArTSeq data underscores the imperative for a more nuanced, molecular-based approach to species identification and classification. This precision is vital for the accurate management of gene banks and the conservation of genetic resources, highlighting the superiority of genotyping technologies over traditional morphological assessments. The alignment of these technologies with conservation and breeding aims promises a more efficient, cost-effective, and accurate preservation and utilization of genetic diversity, which is paramount for addressing the contemporary challenges of climate change, pest resistance, and food security. The combined insights from these investogations offer a compelling narrative of the genetic underpinnings of wheat species that have been central to human civilization suggesting that the future of agriculture may well depend on understanding and leveraging the genetic legacies of the past. In essence, these investigations weave together a comprehensive picture of wheat genetics in regions foundational to agriculture, challenging existing paradigms and offering new pathways for genetic conservation and crop improvement. The detailed exploration of genetic diversity, differentiation, and structure within and between wheat species highlights the importance of molecular data in advancing agricultural sciences. As we stand on the brink of potential agricultural revolutions, these genetic insights serve as a keystone for developing resilient, productive, and sustainable cultivars, thus ensuring food security and ecological balance for future generations.
Conclusion
As a result of the current study, accessions of TU and TB species were clustered together and revealed to be 2 different species with the analysis of the DArTseq and SNP markers. TU and TB species are difficult to distinguish morphologically because they are very close species in terms of their morphological features. Therefore, in addition to the previously performed morphological identifications, there is a need to confirm species identifications using high-resolution genome data obtained by molecular techniques. In line with the results obtained herein, it is obvious that the identification of some accessions should be re-evaluated. According to results of the present research, it is possible to address the possible misidentifications by comparing the ADMIXTURE, ML, UPGMA, and PCoA analysis results. All of the results indicated that accessions 38 and 63 belong to TB and accessions 15, 18, 26, 34 and 35 belong to TU. Accession 16 is close to TU; however, it may be a hybrid species as well. In conclusion, the results herein demonstrated that the DArTseq method, using high genome data, is a fast, low-cost, and high-accuracy method that can be used in species and population discrimination and is an effective tool for GBM.
Data availability
The data that support the findings of this study are available from the authors but restrictions apply to the availability of these data, which were used under permission from Republic of Türkiye Ministry of Agriculture and Forestry for the current study, and so are not publicly available. Data are, however, available from the authors upon reasonable request and with permission from the Ministry of Agriculture and Forestry.
References
Abbasov M, Sansaloni CP, Burgueño J, Petroli CD, Akparov Z et al (2020) Genetic diversity analysis using DArTseq and SNP markers in populations of Aegilops species from Azerbaijan. Genet Resour Crop Evol 67:281–291. https://doi.org/10.1007/s10722-019-00866-7
Alexander HD, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664. https://doi.org/10.1101/gr.094052.109
Allan V, Vetriventhan M, Senthil R, Geetha S, Deshpande S, Rathore A, Kumar V, Singh P, Reddymalla S, Azevedo CV (2020) Genome-wide DArTSeq genotyping and phenotypic based assessment of within and among accessions diversity and effective sample size in the diverse sorghum, pearl millet, and pigeonpea landraces. Front Plant Sci 11:587426. https://doi.org/10.3389/fpls.2020.587426
Baloch FS, Alsaleh A, Shahid MQ, Çiftçi VE, Sáenz de Miera L, Aasim M, Nadeem AM, Aktaş H, Özkan H, Hatipoğlu R (2017) A Whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from central fertile crescent. PLoS ONE 12(1):e0167821. https://doi.org/10.1371/journal.pone.0167821
Brandolini A, Volante A, Heun M (2016) Geographic differentiation of domesticated einkorn wheat and possible Neolithic migration routes. Heredity 117:135–141. https://doi.org/10.1038/hdy.2016.32
Cabi E (2010) Taxonomic revision of the tribe Triticeae Dumortier (Poaceae) in Turkey. Middle East Technical University, Ankara
Dizkırıcı A, Kansu Ç, Önde S (2016) Molecular phylogeny of Triticum and Aegilops genera based on ITS and MatK sequence data. Pak J Bot 48:143–153
Dorofeev VF, Filatenko AA, Migushova EF, Udachin RA, Jakubtsiner MM (1979) Pshenitsa (Wheat). In: Dorofeev VF, Korovina ON (eds) Cultivated flora of the USSR, vol 1. Kolos, Leningrad
Edet OU, Gorafi YSA, Nasuda S, Tsujimoto H (2018) DArTseq-based analysis of genomic relationships among species of tribe Triticeae. Sci Rep 8:16397. https://doi.org/10.1038/s41598-018-34811-y
Fricano A, Brandolini A, Rossini L, Sourdille P, Wunder J, Effgen S, Hidalgo A, Erba D, Piffanelli P, Salamini F (2014) Crossability of Triticum urartu and Triticum monococcum wheats, homoeologous recombination, and description of a panel of interspecific introgression lines. G3 Genes Genomes Genet 4(10):1931–1941
Fufa M, Gedebo A, Letta T, Lule D (2023) Genetic diversity and population structure analysis in tetraploid wheat (Triticum turgidum spp) germplasm from Ethiopia based on DArTSeq markers. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-023-01775-6
Golovnina KA, Glushkov SA, Blinov AG (2007) Molecular phylogeny of the genus Triticum L. Plant Syst Evol 264:195–216. https://doi.org/10.1007/s00606-006-0478-x
Goncharov NP (2011) Genus Triticum L. taxonomy: the present and the future. Plant Syst Evol 295:1–11. https://doi.org/10.1007/s00606-011-0480-9
Goncharov NP, Golovnina KA, Kondratenko EY (2009) Taxonomy and molecular phylogeny of natural and artificial wheat species. Breed Sci 59:492–498
Haas M, Schreiber M, Mascher M (2019) Domestication and crop evolution of wheat and barley: genes, genomics, and future directions. J Integr Plant Biol 61(3):204–225. https://doi.org/10.1111/jipb.12737
Hyun DY, Sebastin R, Lee KJ, Lee GA, Shin MJ, Kim SH, Lee JR, Cho GT (2020) Genotyping-by-sequencing derived single nucleotide polymorphisms provide the first well-resolved phylogeny for the genus Triticum (Poaceae). Front Plant Sci 11:688. https://doi.org/10.3389/fpls.2020.00688
Kilian B, Martin W, Salamini F (2010) Genetic diversity, evolution and domestication of wheat and barley in the fertile crescent. In: Glaubrecht M (ed) Evolution in action: case studies in adaptive radiation speciation and the origin of biodiversity. Springer, Berlin, pp 137–166
Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y et al (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557:424–428. https://doi.org/10.1038/s41586-018-0108-0
R Core Team (2023) R: a language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R-project.org/
Safari Z, Mehrabi AA (2019) Molecular phylogeny of Aegilops L. and Triticum L. species revealed by internal transcribed spacers of ribosomal genes. J Agric Sci Technol 21(3):699–714
Sansaloni C, Franco J, Santos B, Percival-Alwyn L, Singh S (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat Commun 11:4572. https://doi.org/10.1038/s41467-020-18404-w
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Tan K (1985) Triticum L. In: Davis PH (ed) Flora of Turkey and the East Aegean Islands. University of Edinburgh Press, Edinburgh, pp 245–255
Wallace JG, Mitchell SE (2017) Genotyping-by-sequencing. Current protocol in plant. Biology 2:64–77. https://doi.org/10.1002/cppb.20042
Wang X, Hu Y, He W, Yu K, Zhang C, Li Y, Yang W, Sun J, Li X, Zheng F, Zhou S, Kong L, Ling H, Zhao S (2022) Whole genome resequencing of the wheat a subgenome progenitor Triticum urartu provides insights into its demographic history and geographic adaptation. Plant Commun 3(5):200. https://doi.org/10.1016/j.xplc.2022.100345
Zeibig F, Kilian B, Frei M (2022) The grain quality of wheat wild relatives in the evolutionary context. Theor Appl Genet 135:4029–4048. https://doi.org/10.1007/s00122-021-04013-8
Acknowledgements
We would like to express our gratitude to Faheem Shehzad Baloch for his continual support and advice; Hacettepe Plant Evolution Research Laboratory (PERLab) team members, Cansu Kılıç, Özge Çakmak, Ezgi Küçükel, and Veli Deniz Ünlü for assisting with the laboratory work; and Dilek İnceköse Bağlan, Şenay Boyraz Topaloğlu, and the staff of the TSGB for their valuable support. All data generated or analyzed in this study will be available upon request from authors.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). This research was partly funded by TÜBİTAK under Project no: 123R003.
Author information
Authors and Affiliations
Contributions
This manuscript comprised part of the first author’s MSc thesis results, and the second author was the supervisor of the thesis. The study framework was drawn by BTH, experiments and bioinformatic analyzes were performed by AUO. Article writing was done by BTH and contributed by AUO.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Özsoy, A.U., Tarikahya Hacioğlu, B. Genetic diversity of the Turkish accessions of two progenitor species, Triticum baeoticum Boiss. and Triticum urartu Thum. ex Gandil., using DArTSeq markers. Genet Resour Crop Evol (2024). https://doi.org/10.1007/s10722-024-02046-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10722-024-02046-8