
Abstract
Assessing the effectiveness of different molecular markers is essential for identification of appropriate ones for crop improvement and conservation, hence, inter-simple sequence repeat (ISSR) and start codon targeted (SCoT) markers were used for this study. Sixty-six accessions with different genomes obtained from International Transit Center, Belgium, were used for DNA extraction, amplification with ISSR and SCoT markers and agarose gel electrophoresis. The reproducible bands were scored for analyses. We identified high allelic richness of 299 (ISSR) and 326 (SCoT). Polymorphic information contents (ISSR: 0.9225; SCoT: 0.9421) were high but SCoT exhibited higher level of informativeness. The two markers demonstrated high percentage polymorphic loci (ISSR: 91.21–100%; SCoT: 96.97–100%). Other genetic indicators including effective number of alleles, Nei’s genetic diversity, and Shannon information index were higher in SCoT and further elucidated the usefulness of the markers. Intraspecific genetic diversity, interspecific genetic diversity, coefficient of gene differentiation and level of gene flow revealed extensive gene flow and larger variability within the accessions. Both ISSR and SCoT grouped the accessions via dendrogram, biplot and structure analyses. Though the two marker systems varied in their informativeness, they demonstrated high effectiveness in resolving genetic diversity (GD) of the different accessions, with higher efficiency in SCoT markers. Due to higher GD indices exhibited by SCoT, AS is the most genetically endowed one. Our study showed that SCoT markers are more informative than ISSR for GD exploration, assessment and cluster resolution of Musa species, thereby revealing the potential of SCoT markers for improved breeding and conservation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bananas and plantains (Musa spp.), perennial crops, are the favorite fruit crops of the world. They are globally distributed in more than 120 countries where production is estimated at 116 million tonnes (FAOStat 2014, 2019; Chab et al. 2018). Musa spp., originating from South-East/West Oceania zone, possess edible ones including either Musa accuminata Colla (genome A), or a mixture of A and B called M. balbisiana Colla (genome B), or A and S (M. schizcarpa N. W. Simmonds), or A and T (M. textilis Nee) (Perrier et al. 2011; Christelova et al. 2017; Sardos et al. 2018). Bananas and plantains are the highest export fruit crops (Kumar et al. 2014a, b) and rated fourth most important in sub-Saharan Africa (SSA) after cassava, maize and yam (FAOStat 2014). They are good sources of carbohydrates, vitamin C, potassium and sodium (IBA 2007). Many genomic constitutions exist based on their genomic origins with respect to A and B genomes (Simmonds 1966). Different genotypes were derived from M. acuminata (AA) and M. balbisiana (BB) and classified into different genomic constitutions including diploids (AA, AB and BB), triploids (AAA, AAB and ABB) and tetraploids (AAAA, AAAB, AABB and ABBB) (INIBAP 2008; Pollefeys et al. 2004). The East African bananas, mainly desert ones, (AA, AAB, AAA, ABB and AB), the African plantain bananas (AAB) grown mainly in Central, and the West African and the East African Highland Bananas (EAHB: AAA), among others are used for cooking and beer brewing (Karamura et al. 1998). Other subgroups including ‘Mutika’ (formerly known as ‘Mutika-Lujugira’ and currently a well-known EAHB of AAA) and ‘Mchare’ (AA) are grown as major crops in East African countries (Perrier et al. 2011). Six of these genomes (AA, AAA, AB, AAB, ABB and ABBB) are classified as naturally occurring ones based on taxonomic features and ploidy constitutions (Simmonds and Shepherd 1995).
However, these vital crops have become vulnerable to both abiotic and biotic factors depending on their genomic constitutions. Interestingly, it has been reported that genotypes with “B” genome (in particular ABB type) are more tolerant to abiotic and biotic stresses than those solely possessing “A” genome (Tripathi et al. 2007; Vanhove et al. 2012; Davey 2013; Hu et al. 2015). The world population is fast growing, and it is expected to attain over 9 billion by the year 2050 (Ehrlicha and Harte 2015; FAO 2017). Feeding this overwhelming population level is generating much pressure on agricultural crop production (Kastner et al. 2012; Dempewolf et al. 2014; Khoury et al. 2014). To increase productivity of Musa species, it is essential to investigate the degree of relatedness of different genomes and their GD strength for increased tolerance/resistance to different stressors and to meet present and future challenges.
Harnessing genetic diversities and relationships can reveal accessions capable of withstanding the changing environmental factors. This is because crops with narrower range of GD may fail to survive under climatic extremes. Breeders need Musa species with high GD endowment for resistance to abiotic and biotic stressors, but the accurate identification of such unique ones cannot easily be achieved through conventional breeding, due to the complicated genetic system of Musa species. It is possible by complementing it with informative and functional gene-based molecular markers that are not influenced by changes in climatic factors (Martínez et al. 2006). Different molecular markers (with emphasis on gel-based molecular marker techniques) including random amplified polymorphic DNA (RAPD) marker (Bhat et al. 1995; Ude et al. 2003a, b; Lamare and Rao 2015), restriction fragment length polymorphism (RFLP) (Bhat et al. 1995), simple sequence repeat (SSR) (Perrier et al. 2011; Hippolyte et al. 2012; Christelova et al. 2017), sequence-related amplified polymorphism (SRAP) (Youssef et al. 2011), inter-simple sequence repeats (ISSR) (Godwin et al. 1997; Lamare and Rao 2015), directed amplified minisatellite DNA (DAMD) (Lamare and Rao 2015) and amplified fragment length polymorphism (AFLP) (Ude et al. 2003a, b; Bhat et al. 2004) have been used in Musa species. Considering the “indirect random markers” such as RAPDs, ISSRs, or SSRs (Andersen and Lübberstedt 2003) only without regards to other gene-based markers, limits the expected progress in germplasm improvement, utilization and conservation. Informative molecular markers can effectively exploit GD and relatedness inherent in Musa species and their wild relative species for genetic improvement as earlier reported in other crops (Hajjar and Hodgkin 2007; Ford-Lloyd et al. 2011). In addition, conserving GD of domesticated and wild crop relative species via the use of functional gene-based markers can provide a strong basis for the sustainable use of these species. Knowledge of the GD within different accessions of Musa species possessing diverse genomic constitutions can facilitate the efficiency of banana and plantain breeding programs. Comparative utilization of ISSR and Start codon targeted (SCoT) markers has become very important to identify more informative and better resolving markers for Musa species. Inter-simple sequence repeat markers which are arbitrary in nature target variable genomic loci for amplification of DNA segments present between two similar microsatellite regions that are opposite with each other (Zietkiewicz et al. 1994), while SCoT markers were developed from the conserved regions of the genomes across different plant species and they possess longer primer lengths and high annealing temperatures (Collard and Mackill 2009). Although, ISSR and SCoT markers have been found useful in various crops for GD studies because of their high reproducibility and efficiency (Godwin et al. 1997; Galvan et al. 2003; Cao et al. 2006; Collard and Mackill 2009; Guo et al. 2012; Hamidi et al. 2014; Etminan et al. 2016; Igwe et al. 2017), SCoT is yet to be utilized in bananas and plantains. To the best of our knowledge, comparative assessment of effectiveness of ISSR and SCoT markers has not been utilized in bananas and plantains with different ploidy or genomic constitutions to harness their usefulness in assessing GD, cluster resolution and population structure. Therefore, the objectives of this study were to: (1) study the GD and cluster resolution among the 66 accessions of Musa species using ISSR and SCoT markers; (2) compare the effectiveness of ISSR and SCoT markers in assessing GD and population structure of 66 accessions of Musa species with different genomes.
Materials and methods
Sample collection and DNA extraction
Sixty six (66) accessions of bananas and plantains from different genomes, namely, AA, AAA, AAAA, AAB, BB, AB, ABB, AAAB and AS, as well as other three diploid accessions (Musa beccarii N. W. Simmonds, M. coccinea Andrews, and M. textilis) were obtained from the International Transit Center (ITC), Kasteelpark Arenberg 13 bus 2455, 3001 Leuven, Belgium (Supplementary file 1 Table S1). Thirty-two (32) out of 66 were obtained as tissue cultured plantlet materials, each in five replicates, and were grown and maintained at the screenhouse of the Department of Natural Sciences, Bowie State University, while the remaining 34 were obtained in lyophilized form. Approximately 100 mg and 120 mg were, respectively weighed from young fresh and lyophilized leaves of Musa species for DNA extraction using Cetyltrimethylammonium bromide (CTAB) method (Abarshi et al. 2010) with a little modification by using ratio of 241:1 of chloroform and isoamyl alcohol, respectively without phenol.
Polymerase chain reaction and agarose gel electrophoresis
Polymerase chain reaction (PCR) amplification was performed in volume of 25µL which consisted of 2.0 µL 100 ng DNA, 5.0 µL of 5 × Green GoTaq Buffer (Promega Corporation, Madison, USA), 2.0 µL of 2.5 mM dNTPs (Bioline, Massachusetts, USA), and 0.2 µL GoTaq DNA polymerase (5 U/µL) (Promega Corporation, Madison, USA), 1.0 µL of 10 µM each of ISSR or SCoT primer and 14.80 µL of 500 mL diethyl pyrocarbonate (DEPC)-treated water (Invitrogen, Carlsbad, CA, USA). The list of ISSR and SCoT primers, their sequences, Guanine-Cytosine (GC) content, annealing temperatures and sources were presented (Table 1). The PCR amplification profile was run for 35 cycles, each consisting of a denaturation step at 94 °C for 5 min, followed by annealing at 35–40 °C for 35 s, an extension step at 72 °C for 1 min, and a 10 min final extension at 72 °C using a Bio-Rad T100 Thermal cycler (Bio-Rad Laboratories Inc. Singapore). Ten (10) µL of the PCR reaction products were resolved with 1.5% agarose gel containing 0.5 mg/mL ethidium bromide and photographed using Aplegen Omega Lum G gel documentation system (Minnesota 55303, USA).
Data analyses
Data matrices of ISSR and SCoT marker profiles were generated by scoring (1) for presence and (0) for absence of individual allele. The generated data matrices were used for computing GD, allele frequency and polymorphic information content (PIC) using PowerMarker (Version 3.25). Analyses of number of polymorphic loci (NPL), percentage polymorphic loci (PPL), effective number of alleles (Ne), Nei’s gene diversity, Shannon’s information index (I), and population structure (total gene diversity or intraspecific GD, Ht; gene diversity within accessions or interspecific GD, Hs; coefficient of gene differentiation, GST; and level of gene flow, Nm) of the accessions were analyzed using POPGENE software version 1.32 (Yeh and Boyle 1997). Brown’s analysis for multilocus associations among and within populations was also performed with the POPGENE program. The multilocus association variances were partitioned into single-locus and two-locus components (Brown and Feldman 1981). Dendrogram reconstruction using Unweighted Pair Group Method with Arithmetic Mean (UPGMA Pakseresht et al. 2013), SAHN clustering and dissimilarity index in Jaccard’s option was conducted using NTSYSpc software version 2.02 (Rohlf 2000). Principal component analysis (PCA) and population structure of ISSR and SCOT datasets were computed as Discriminant analysis of principal component (DAPC) using Bayesian information criterion (BIC) as described by Jombart (2008), Jombart et al. (2010), Jombart and Collins (2015) in the Adegenet package v.2.1.2 in R (R Development Core Team 2015). Biplot function in R (R Development Core Team 2015) was used to plot the first two PCAs with individual points coloured and shaped according to cluster membership identified through DAPC.
Results
Allelic variation, gene diversity and polymorphic information content obtained using inter-simple sequence repeat and start codon targeted markers
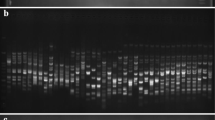
Nine out of the 13 primers of ISSR tested were replicated and found reproducible and scorable, while eight out of 19 were selected from SCoT as in some of the representatives of the gel images (Fig. 1a, supplementary file 2 Figs. S1a and S1b; Fig. 1b, supplementary file 2 Figs. S1c and S1d). Each of the representatives of the gel images had unique banding patterns across some of the accessions. For the ISSR, the reproducible ones were UBC808, UBC825, UBC827, UBC835, UBC840, UBC901, UBC811, UBC812, and UBC840B, while SCoT consisted of SCoT16, SCoT2, SCoT20BM, SCoT33, SCoT36, SCoT20, SCoT22, and SCoT26. A total of 299 numbers of alleles were generated from the reproducible ISSR markers, while SCoT yielded 326 (Table 2). The major allele frequency in ISSR was 0.1424, while SCoT had 0.1717. The gene diversities obtained with ISSR and SCoT were 0.9282 and 0.9432, respectively. Polymorphic information content values of 0.9225 (ISSR) and 0.9421 (SCoT) were obtained. The ISSR primers UBC808, UBC825, UBC827, UBC835, UBC840, UBC901, UBC811 and UBC812 were polymorphic, while UBC840B was monomorphic. The PIC values from the ISSR primers were ranked in a descending order as UBC808 > UBC811 > UBC827 > UBC825 > UBC835 > UBC901 > UBC840 > UBC812 > UBC840B. The identified polymorphic SCoT primers were SCoT16, SCoT2, SCoT20BM, SCoT33, SCoT36, SCoT20 and SCoT22, while SCoT26 was monomorphic. The PIC values from the SCoT were SCoT20BM > SCoT2 > SCoT20 > SCoT36 > SCoT33 > SCoT16 > SCoT22 > SCoT26. Allelic scores, counts and frequencies obtained using ISSR and SCoT marker systems were informative with variable allelic count ranges (ISSR: 1–21; SCoT: 1–24) and frequencies (ISSR: 0.0152–0.3182; SCoT: 0.0152–0.3636) (supplementary file 3 Table S2; supplementary file 4 Table S3).

Amplification profile of 66 banana and plantain samples using UBC825 (a) and SCoT36 (b) primers: a = 100 bp step DNA ladder and b = 1 kb DNA ladder; red arrows indicate unique allelic/banding patterns in some of the accessions; sample order (1–66 from left to right): 1 = Fougamou 1, 2 = Obino I'Ewai, 3 = Calcutta 4, 4 = Improved Lady Finger, 5 = Blue Torres Strait Island, 6 = Silk, 7 = Truncata, 8 = Cardaba, 9 = Lidi, 10 = Pelipita, 11 = Pelipita Manjoncho, 12 = Lai, 13 = Higa, 14 = Pisang Keling, 15 = Pisang Lawadin, 16 = Balonkawe, 17 = Gros Michel, 18 = Green Red, 19 = Plantain no.3, 20 = Pata, 21 = Chinese Cavendish, 22 = Dwarf Parfitt, 23 = Hochuchu, 24 = Umalag, 25 = Hsein Jen Chiao, 26 = Mons Mari (Pedwell), 27 = Lady Finger (Nelson), 28 = Pisang Rajah (South Johnstone), 29 = Tani, 30 = Pisang Lilin, 31 = Poteau Geant, 32 = Pisang Klutuk Wulung, 33 = Garbon 2, 34 = Zebrina (G.F), 35 = Khae (Phrae), 36 = Dole, 37 = Wompa, 38 = Pisang Palembang, 39 = Pisang Awak, 40 = Williams (Bell, South Johnstone), 41 = Plantain No.17, 42 = Kluai Tiparot, 43 = Tiau Lagada, 44 = Niyarma Yik, 45 = Selangor, 46 = Long Tavoy, 47 = Malaccenesis, 48 = Figure Pomme Geante, 49 = Highgate, 50 = Borneo, 51 = Honduras, 52 = Pome, 53 = Kunnan, 54 = Musa beccarii N. W. Simmonds, 55 = Musa coccinea Andrews, 56 = JD Yangambi, 57 = Musa textilis, 58 = Tomolo, 59 = Pisang Berlin, 60 = FHIA-23, 61 = No.110, 62 = Dwarf Cavendish, 63 = SH-3436–6, 64 = Lal Velchi, 65 = Madang and 66 = FHIA-21 (#68)
Genetic diversity within inter-simple sequence repeat and start codon targeted markers
The identified NPL and PPL from the nine reproducible set of primers of ISSR markers were 47–66 and 91.21–100, respectively, while the eight reproducible set of primers of SCoT markers yielded 65–66 (NPL) and 96.97–100 (PPL) (Table 3). Based on the GD richness, 8 of the ISSR primers exhibited 91.21–100% polymorphisms, while seven of the SCoT primers showed 96.97–100% polymorphism. Within the nine primers of ISSR markers, Ne, H and I values and their standard deviations ranged from 1.3590 ± 0.2986–1.7802 ± 0.2219, 0.2269 ± 0.1739–0.4272 ± 0.0925 and 0.3491 ± 0.2531–0.6132 ± 0.1149, respectively, while the Ne, H and I values and their standard deviations from SCoT markers were 1.4447 ± 0.1775–1.8249 ± 0.2231, 0.2848 ± 0.1209–0.4411 ± 0.0917 and 0.4505 ± 0.1504–0.6288 ± 0.1064, respectively.
Genetic diversity of different genomic constitutions of bananas and plantains as revealed by Inter-simple sequence repeat and Start codon targeted markers
Within the Musa species of diverse genomic constitutions assessed with ISSR and SCoT marker systems, variable GD indices were identified in both ISSR and SCoT (Table 4). Assessing the genomes of AA, AAA, AAAA, AAAB, AAB, BB, and AB, ISSR produced genetic parameters that were different from those obtained with SCoT markers. For the wild and other diploid (M. textilis M. coccinea, and M. beccarii) accessions studied with ISSR, different GDs (Ne = 1.2941, H = 0.2273, I = 0.3878) were detected in relation to those from SCoT (Ne = 1.8312, H = 0.4539, I = 0.6463).
The GD endowment in an ABB genome was found to be the highest, with the values of Ne, H and I as 1.6310, 0.3801 and 0.5657, respectively, using ISSR, while SCoT identified AS to be the highest (Ne = 1.8312, Ne = 0.44539, I = 0.6463). On the other hand, the GD in the AS accession was lowest (Ne = 1.2941, H = 0.2273, I = 0.3878) with ISSR, while SCoT determined textilis as the lowest judging from the values of GD (Ne = 1.4382, H = 0.3047, I = 0.4826). The GD indicators identified in these variable genomic (ploidy) groups were ranked: ABB > AAB > AAAB > BB > AAA > AA > coccinea > AAAA > textilis > AB > beccarii > AS from high to low based on polymorphic loci of the selected ISSR, while SCoT ranked them as AS > AAA > coccinea > AAB > AAAA > AA > beccarii > ABB > AB > BB > AAAB > textilis from high to low. From the ISSR, the overall mean values of Ne, H and I and their respective standard deviations across the diverse genomic constitutions were 1.5474 ± 0.1509, 0.3476 ± 0.0649 and 0.5296 ± 0.07460, respectively, while SCoT yielded 1.6440 ± 0.1749 (Ne), 0.3841 ± 0.0737 (H) and 0.5694 ± 0.0858 (I).
The assessment of genetic variations within and among the different accessions revealed that the values of Ht, Hs, GST and Nm were distinctly different based on the two markers (Table 5). For the ISSR, there were different values of Ht (0.2273–0.3837), Hs (0.2076–0.3516), GST (0.0537–0.1180) and Nm (5.2826–12.4521) across the entire accessions relative to those (Ht = 0.3047–0.4539; Hs = 0.0323–0.4222; GST=0.0630–0.1699; and Nm = 2.4421–14.7687) that were generated with SCoT. With ISSR, the overall mean values of Ht, Hs, GST and Nm were 0.3476 ± 0.0042, 0.3229 ± 0.0036, 0.0709 and 6.5531, respectively, while 0.3841 ± 0.0054, 0.3452 ± 0.0044, 0.1013 and 4.4377 were obtained with SCoT. Using ISSR markers, the GST of 7.09% was recorded as the total genetic divergence among the accessions and 92.91% was found within the accessions, while SCoT identified 10.13% as the total genetic divergence among the accessions, and 89.87%, within the accessions.
Dendrogram analysis of different genomic constitutions of Musa species as revealed by inter-simple sequence repeat and start codon targeted markers
A dendrogram analysis generated five major groups at a similarity index of approximately 0.80 using ISSR markers (Fig. 2). Group I consisted of ‘Fougamou 1’, ‘Pisang Palembang’, ‘Improved Lady Finger’ and ‘Silk’, with AAB-genome accessions dominating the group. Group II contained ‘Pisang Rajah’ (South JohnStone) that had AAB genome. In group III, five subclades, SCI, SCII, SCII, SCIII, SCIV and SCV were respectively identified. For SCI, accessions such as ‘Obino I’Ewa’, ‘Higa’, ‘Garbon 2’, ‘Plantain no17’, ‘Calcutta 4’, ‘Cardaba’, ‘Lidi’, ‘Pisang Lilin’, ‘Lai’ and ‘Green Red’ were found with AA/AAA genome as the most abundant ones compared to AAB. ‘Garbon 2’ (AAB), ‘Plantain no17’ (AAB) got closely clustered. Similar clustering was also observed between ‘Lidi’ (AA) and ‘Pisang Lilin’ (AA); ‘Lai’ (AAA) and ‘Green Red’ (AAA) within SCI. Exceptions were found between ‘Obino I’Ewa’ and ‘Higa’ that had different genome groups but still tightly clustered together and this similar grouping was also detected between ‘Calcutta 4’ and ‘Cardaba’. In SCII, only ‘Truncata’ (AA) was found even though it had the same genome group possessed by some accessions in SCI, while SCIII had ‘Gros Michel’, ‘Chinese Cavendish’, ‘Honduras’, ‘Umalag’, ‘Hsein Jen Chiao’, ‘Dwarf Parfitt’, ‘Mons Mari’ (Pedwell) and ‘Lady Finger Nelson’ that were mostly triploid A (AAA), except ‘Lady Finger Nelson’ (AAB) and ‘Honduras’ (BB).

Dendrogram of 66 accessions of Musa species generated using inter-simple sequence repeat markers
In SCIV, accessions of different ploidy groups including ‘Pisang Keling’, ‘Figure Pome Geante’, ‘Williams’ (Bell, South Johnstone), ‘Highgate’, ‘Selangor’, ‘Long Tavoy’, ‘Malaccenesis’, ‘J.D Yangambi’ and ‘Pome’ were identified, while ‘Khae’ (Phrae), ‘Wompa’, ‘Tjau Lagada’, ‘Tomolo’, ‘Pisang Berlin’, ‘No.110’, ‘SH-3436-6’, ‘Borneo’ and ‘FHIA-23’, which were dominated by A genome, were detected in SCV subclade. In group IV that got descended from M. coccinea accession, mixed triploids (AAA and AAB) and A diploid were revealed in SCI, while both diploid and tetraploid A genome dominated SCII. Triploid accessions ‘Pisang Lawadin’, ‘Plantain no3’, ‘Pata’, ‘Poteau Geante’ and ‘Balonkawe’ with mostly AAB genome clustered in SCI. Furthermore, accessions ‘Zebrina-G.F’, ‘Niyarma Yik’, ‘Honduras’, ‘Kunnan’, M. beccarii (diploid), ‘Dwarf Cavendish’, ‘Lal Velchi’, M. textilis (diploid) and ‘Dole’ were contained in SCII and this sub-group contained AA, BB,AB,ABB and AAA. In group V, accessions of different ploidy groups including ‘Blue Torres Strait Island’, ‘Pelipita’, ‘Pelipita Manjoncho’, ‘Tani’, ‘Pisang Klutuk Wulung’, ‘Pisang Awak’, ‘Khrae Tiparot’, ‘Madang’ and FHIA-21 (#68) clustered together but ‘Madang’ and FHIA-21 (#68) were more distantly isolated.
A dendrogram analysis with SCoT markers identified five major groups at similarity index of 0.75 (Fig. 3). For group I, three sub-clades of SCI, SCII and SCIII were detected. In SCI, ‘Fougamou1’, ‘Pelipita Manjoncho’, ‘Plantain no17, ‘Pome’, ‘Zebrina G.F’, ‘Wompa’, ‘Williams’ (Bell, South Johnstone), ‘Silk’, ‘Pisang Palembang’ and ‘Figure Pome Geante’ with mixed diploids (AA, AS) and triploids (AAA, ABB, AAB) as their respective genomic groups were revealed. The ‘Silk’, ‘Pisang Palembang’ and ‘Figure Pome Geante’ were more isolated in this group. For SCII, accessions ‘Calcutta 4’, ‘Pisang Lawadin’, ‘FHIA-23’, ‘No.110’, ‘JD-Yangambi’, ‘Plantain no3’, ‘Lady Finger Nelson’, ‘Gros Michel’, ‘Highgate’, ‘Mons Mari’ (Pedwell), ‘Pisang Awak’, ‘Selangor’, ‘Long Tavoy’, ‘Tjau Lagada’, ‘Tomolo’, and ‘Pisang Berlin’. In this subclade, ‘Calcutta 4’ and ‘Pisang Berlin’ were genetically isolated from other accessions. In SCIII, diverse genomic compositions (AA, AAA, AAAA, ABB, AAB) associated with different accessions clustered together. The identified accessions included ‘Improved Lady Finger’, ‘Pelipita’, ‘Poteau Geante’, ‘Garbon 2’, ‘Chinese Cavendish’, ‘Hsein Jen Chiao’, ‘Hochuchu’, ‘Lidi’, ‘Borneo’, and ‘Pisang-Lilin’. Chinese Cavendish’, ‘Hsein Jen Chiao’ and ‘Hochuchu’ each had AAA and got closely grouped together and the same clustering was applied to ‘Lidi’, ‘Borneo’, and ‘Pisang-Lilin’ that AA.

Dendrogram of 66 accessions of Musa species generated using Start codon targted (SCoT) polymorphic markers
In group II, two subclades, SCI and SCII originating from ‘Cardaba’ (ABB) and ‘SH-3436-6’ (AAAA) were formed. Within the SCI, ‘Obino I’Ewa’, ‘Blue Torres Strait Island’, ‘Green Red’, ‘Balonkawe’, ‘Pata’, ‘Dwarf Parfitt’, ‘Dole’, ‘Niyarma Yik’, ‘Dwarf Cavendish’, ‘Malaccenensis’, ‘Truncata’, M. textilis (textilis), ‘Lal Velchi’ and M. beccarii (beccarii) grouped together, with ‘Truncata’, M. textilis (textilis), ‘Lal Velchi’ and M. beccarii (beccarii) demonstrating much distant separation from other accessions. Sub-group SCII consisted accessions with B genome as the most dominant, but ‘Madang’ (AA) and FHIA-21 (#68) (AAAB) were distantly related. In group III, only ‘Khae (Phrae)’, ‘Honduras’ and ‘Kunnan’ were identified. Groups XIV and V had ‘Lai’ and ‘Umalag’ each with AAA genome.
Principal component analysis, biplot and population structure of different genomic constitutions of Musa species as revealed by inter-simple sequence repeat and start codon targeted markers
PCA was computed through DAPC to identify association among the accessions with variable genomic groups, with the first three PCs showing 28.5% and 27.8% variance in ISSR and SCoT marker datasets, respectively. Biplots revealed clustering pattern with PCA 1 and PCA 2 explaining about 12.3% and 9.3% variance in ISSR and 12.5% and 8.5% in SCOT, respectively (Fig. 4). Cluster 1 of the population structure obtained with ISSR markers, contained accessions with three variable genomes (BB, ABB, AAB), while cluster 1 in SCoT had four (AA, AAA, AAB, AAAA) (Fig. 5). For cluster 2 in ISSR, nine genomic groups including wild diploids (AA, BB, AB, ABB, AAA, AAAA, AAAB, AAB, AS, wild diploids), were detected and seven with wild diploids (AA, BB, ABB, AAA, AAAB, AAB, AAAA, wild diploids) in SCoT. In cluster 3, four genomic groups (AA, AAA, ABB, AAB) and seven with wild diploids (AA, AB, AAB, ABB, AAA, AAAA, AS, wild diploids), were respectively identified with ISSR and SCoT marker systems.

Principal component analysis of 66 Musa species accessions assessed with Inter-simple sequence repeat (a) and Start codon targeted (b) marker systems

Population structure of 66 Musa species accessions assessed with Inter-simple sequence repeat (a) and Start codon targeted (b) markers
Comparison of data generated from inter-simple sequence repeat and start codon targeted markers across 66 different genomes of banana and plantain accessions
A total number of 299 alleles (14–57 per primer) were obtained with ISSR markers, while 326 alleles (26–60 per primer) were identified with SCoT (Table 6). Other genetic indices including mean major allele frequency, allelic count, gene diversity, PIC, NPL, PPL, Ne, H, and I were distinctly and differently revealed by the two marker systems. The overall mean values of Ne, H and I in ISSR were 1.5474 ± 0.1509, 0.3476 ± 0.0649, and 0.5296 ± 0.07460, while SCoT had 1.6440 ± 0.1749, 0.3841 ± 0.0737, and 0.5694 ± 0.0858. Also, the values of Ht, Hs, GST and Nm parameters varied between ISSR and SCoT markers. Overall average values of Ht, Hs, GST and Nm in ISSR were 0.3476 ± 0.0042, 0.3229 ± 0.0036, 0.0709 and 6.5531, whereas 0.3841 ± 0.0054, 0.3452 ± 0.0044, 0.1013 and 4.4377 were, respectively, obtained with SCoT marker systems. Nine and 15 major groups with various subclades at similarity indices of 0.81 and 0.77 were, respectively obtained from the dendrogram analysis of the 66 accessions using ISSR and SCoT data.
To further evaluate the genetic structure associated with the accessions, analysis of multilocus associations was applied to both the ISSR and SCoT data (Table 7). For the entire population of the Musa accessions, the mean gene diversity (MH) of single-locus components obtained with ISSR markers accounted for 13.4% (with variance of diversity, VH = 0.7882), while SCoT accounted for 14% (with VH = 0.6357). For the populations, the obtained Wahlund’s effect (WH) indices were 0.4951 with ISSR and 0.6275 with SCoT markers. Considering the two-locus effect, the mean disequilibria (MD) obtained with ISSR markers (289.0036) varied with those of SCoT (194.3081). The Wahlund’s effect (WC) values in two-locus effect in the populations were 11.8337 (ISSR) and 14.8983 (SCoT). Other parameters including interactions between MD and WC (abbreviated as AI), variance of disequilibrium (VD), covariance of interactions (CI), total variance (σ2t) and average variance (σ2a) in the populations varied between the two markers.
Discussion
Genetic diversity of banana and plantain is important to the development of sound Musa breeding programs, especially now that the issue of climate change is globally threatening crop productivity and yields (Ortiz 2000; Khayat and Ortiz 2011). Assessing the GD of these vital crops using informative and gene-based molecular markers can readily facilitate the exploration of Musa accessions for selection and integration into breeding programs for crop improvement and conservation. Comparative application of different molecular markers to explore GD or allelic richness of crops has provided a detailed and more informative characterization as well as the choice of the best approach of classification available to researchers than a single marker system (Souframanien and Gopalakrishna 2004; Scariot et al. 2007; Lamare and Rao 2015; Igwe et al. 2017). To the best of our knowledge, this is the first report of utilizing and comparing the effectiveness of ISSR and SCoT markers in accessing GD and population structure of accessions of Musa species with different genomic compositions. Our results demonstrate high potential in assessing GD of the accessions as exhibited by the unique banding patterns or allelic variations, high GD and population parameters including NPL, PPL, PIC, Ne, H, I, Ht, Hs, GST, among other essential indices generated with these markers. Reports show that accessions with the high GD of neutral markers and alleles might be suitable candidates for high adaptive variation, fitness, and conservation (Van Zonneveld et al. 2012; Ilves et al. 2013). In addition, it has been reported that allelic richness is an indicator of GD and it is mostly utilized to assess molecular markers and to identify crops for conservation and breeding purpose (Vinceti et al. 2013).
Both marker systems in this study showed high values for the total number of alleles, mean numbers of alleles, major allele frequency, allelic counts, GD, PIC, and other genetic indicators; but these crucial parameters were higher in SCoT than those obtained with ISSR markers. This demonstrates that SCoT markers may be more effective and efficient in dissecting GD, and relationships among the accessions. The total number of alleles or bands and their averages (no of alleles/bands, n = 299; average, av = 36.5) obtained from ISSR are higher than the ones earlier reported in 21 and 25 accessions of Musa species with mixed genomic constitutions (Lamare and Rao 2015; Silva et al. 2016), but lower than the ones (n = 326; av = 41) from SCoT. The differences could be due to the nature and number of accessions assessed as well as the loci of the markers. The major allele frequency of 0.1424 from ISSR was lower than 0.1717 from SCoT and this is expected since SCoT markers are derived from the functional section of genomes and more informative (Collard and Mackill 2009). Polymorphic information contents from the two maker techniques are quite high, thereby demonstrating the informative nature of these marker systems but SCoT displayed the higher value of PIC. Different but lower values of PIC were shown in 21 Musa species that consists of triploid and tetraploid accessions (Silva et al. 2016) and 25 variable accessions (Lamare and Rao 2015). Efficiencies of the two marker systems have been exploited in another plant in which SCoT was considered more efficacious than ISSR, but the identified PIC of SCoT is slightly higher (Igwe et al. 2017). The noted discrepancy could be associated with the mixed ploidy nature and number of the accessions used in this study.
The mean major allele frequencies produced by the two markers, ISSR and SCoT from the 66 accessions are high especially the one from SCoT, but lower than the one obtained using SSR markers across 695 accessions, including 327 diploids, 363 triploids and 5 tetraploids (Hippolyte et al. 2012). This could be due to the large difference in the number of accessions analyzed with the SSR markers.
The NPL is used to evaluate the efficiency of primers in dissecting the GD among individuals of accessions (Luz et al. 2015) and this is consistent with our study that generated high NPL with ISST (524) and SCoT (567). Interestingly, both markers produced high PPL varying from 91.21 to 100%, with SCoT demonstrating higher efficiency by possessing a higher range of PPL (96.97–100%) than ISSR (91.21–100%). In a study involving polyploidy plant, durum wheat (Triticum turgidum var. durum) that is similar to Musa species, high polymorphisms were recorded using ISSR (98.70%) and SCoT (100%) markers (Etminan et al. 2016). Other researchers identified varying rates of polymorphisms of 85.1% (Ying et al. 2011), but slightly in agreement with 97.5% (Silva et al. 2016).
The overall mean values of Ne, H and I from ISSR (Ne = 1.5474 ± 0.1509, H = 0.3476 ± 0.0649, I = 0.5296 ± 0.07460) are lower than those from SCoT (Ne = 1.6440 ± 0.1749, H = 0.3841 ± 0.0737, I = 0.5694 ± 0.0858) and these high parameters revealed intermediate degrees of GD inherent in the studied accessions. Silva et al. (2016) identified lower numbers than the ones obtained in this study possibly due to the higher number of mixed ploidy accessions used. Also, Lamare and Rao (2015), reported lower mean values of Ne, H and I as 1.589 (± 0.331), 0.338 (± 0.160) and 0.499 (± 0.215) in 25 accessions using RAPD, ISSR and DAMD markers. The AAB genome was identified as the most genetically diverse with ISSR technique, and AS with SCoT. The difference could be as a result of the degree of informative loci accessed by the two marker systems, but SCoT seem to be more accurate for identifying a wild accession as the most genetically diverse. This is because continuous selection decreases effective population size and increases genetic drift in the course of domestication (Doebley et al. 2006; Tang et al. 2010). It has also been reported in various crops that inbreeding and extensive selections narrow the germplasm genetic base and reduce GD richness, especially during the domestication process (Voisin et al. 2005; Doebley et al. 2006).
The population diversity indices like allelic richness (Na and Ne), Nm and GST assess the genome compositions of a population. Nei’s genetic differentiation among the accessions ranges from zero to one, with high values representing a large amount of variation among different accessions or populations. We identified GST values of 0.0709 (7.09% for total genetic divergence among the accessions and 92.91% for within the accessions) with ISSR and 0.1013 (10.13% for total genetic divergence among the accessions and 89.87% for within the accessions) with SCoT, indicating that majority of the variation is within the populations of the accessions whereas minor polymorphism was among the accessions as reported in Durum wheat (Etminan et al. 2016). The identified GST in this study is lower than the one obtained by Lamare and Rao (2015) using combined GST values from three markers (RAPD, ISSR and DAMD); Ge et al. (2005) using wild M. balbisiana with cpDNA PCR–RFLP; and Mukunthakumar et al. (2013) that analyzed wild banana populations with RAPD markers. This finding could be linked to the effect of summation of GST values from three marker systems, and the use of wild populations that have proved to be more genetically diverse than the cultivated ones (Zawko et al. 2001). The higher proportion of the genetic polymorphisms within the accessions and lower degree of genetic differentiation among the populations using the two markers (ISSR: 7.09% for among and 92.91 within accessions; SCoT: 10.13% for among and 89.87 within the accessions) corroborate with other studies (Lamare and Rao 2015). It has been reported that most of the out-breeding accessions or species usually have higher GD within populations and low diversity among populations (Hamrick and Godt 1996; Hogbin and Peakall 1999; Zawko et al. 2001). Estimate of gene flow has been categorized as low (Nm < 1), moderate (Nm > 1) and extensive (Nm > 4) in nature (Kumar et al. 2014a, b). The values of Nm from the ISSR and SCoT data in the present study are high since Nm > 4 in both markers, indicating a high and extensive profile but a higher value was obtained with ISSR. This high level of gene flow demonstrates lack of habitat fragmentation, which could be natural or human disturbance that determine the rate of gene flow as earlier reported (Padmesh et al. 2012).
Dendrogram analysis revealed five major groups from ISSR and SCoT data, respectively with different subclades in 66 sets of accessions. This variation in the number of sub-clusters obtained from the both markers could be due to the more effectiveness of the SCoT markers in targeting different loci, which in turn resulted to existence of some accessions in separate groups instead of aligning with their respective larger groups. Most of the genomic groups were well resolved by the markers. For instance, in group I of ISSR, the AAB triploids dominated the group. In group III with several subclades (SCI-SCV), each sub-cluster resolved the accessions based on genomic constitutions as obtained in SCI that had most A genome (AA/AAA) compared to those that had AAB. However, the ‘Gros Michel’ and ‘Chinese Cavendish’ with triploid AAA clustered closely in SCII of group III with AAA, AA and AAB and this relatedness is not unusual since they share AA genomic group as earlier reported (Perrier et al. 2011; Rotchanapreeda et al. 2015). In group IV, mixed triploids (AAA, AAB) and diploid A were found in SCI, while diploid and tetraploid A genomes were more predominant in SCII. SCIII had accessions with triploid A (AAA) genome as the most frequently occurring, except AAB (‘Lady Finger Nelson’), and BB (‘Honduras’) that were also detected in the same sub-group. Furthermore, AAA and AAB dominated the group and this over-representation of these two genomes has been reported using SSR markers (Hippolyte et al. 2012). Within SCIV of group III of the ISSR dataset, ‘Figure Pome Geante’ (AAB), a donor of the balbisiana genome, clustered closely to ‘Pisang Keling’ (AAB) but distantly isolated from ‘Pome’ (AAB), while ‘Williams’ (Bell South Johnstone) and ‘Highgate’ with AAA genome are tightly grouped together compared to others (‘Selangor’ and ‘JD-Yangambi’) with the similar genome. This similar heterogenomic grouping was reported in Musa accessions analyzed with AFLP markers (Ude et al. 2002) and SSR markers (Sardos et al. 2018). It has also been reported that the triploid AAB had vital contribution of the genome from M. acuminata Colla (Simmonds 1966). In SCI of group I of SCoT marker dataset, genomes of AA, AS, and AAAA were found with AA dominating the group. Group II contained all the plantain genomes (AAB and ABB) but ‘Balonkawe’ is more distantly related to other accessions. The grouping of ‘Madang’ (AA) and ‘FHIA21’-(68) (AAAB) in SCII of group II is in order since crossing between AAB Plantain and AA could produce AAAB genomic constitution (Baurens et al. 2018). For SCoT, most of the groups clustered mostly diploids, triploids and tetraploids including ABB, AAB, AAB/AAAA, ABB/AAA, AA, and ABB/AAA/AA, and these similar heterogeneous grouping patterns were reported using SSR markers (Hippolyte et al. 2012; Mukunthakumar et al. 2013). In SCI of group II, the ABB genome are closer to the AAB than the BB in SCII of the same group as previously reported using AFLP (Ude et al. 2002). The clustering of diploids (AA/BB) and triploids (AAB/ABB) in SCII of group II is expected due to possible harboring of AA genome (Raboin et al. 2005). Other groups from SCoT with singleton accessions include groups III, IV and V that possess AA, AAA and AAA genomic groups, respectively, except in ‘Kunnan’ (AB) and ‘Honduras’ (BB). The detected groups with ISSR and SCoT are lower than the ones reported using SSR markers in 48 accessions (Hippolyte et al. 2012) possibly due to the different nature of the accessions assessed. In ISSR, two diploid accessions (M. beccarii and M. textilis) got clustered closely with known diploid accessions of ‘Kunnan’ (AB) and ‘Lal Velchi’ (BB) confirming their existing diploid genomes, while the third one (M. coccinea) was genetically isolated in a separate group. Surprisingly, both ISSR and SCoT markers clustered diploid (‘Madang’: AA) and tetraploid (‘FHIA21’-(68): AAAB) together and this demonstrates the effectiveness of the resolution of the markers as well as possible hybridization followed by a significant role of AAB in AA genome (Simmonds 1966). However, SCoT resolved them differently by grouping M. textilis and M. beccarii together with ‘Lal Velchi’ accession that has diploid BB genome, which implies that the genomic group of M. textilis and M. beccarii accessions could be BB. Also, M. coccinea is genetically isolated and did not cluster with either M. textilis or M. beccarii accession but with the other triploids. The discrepancies in the clustering could possibly be linked to misclassification resulting from sample mislabeling and handling errors. Similar misclassification was previously reported in accessions Gebi (ITC0877) and Mwitu Pemba (ITC1545) that were previously regarded as triploids but subsequently confirmed to be diploids (Hippolyte et al. 2012).
PCAs and biplots of the accessions with the two marker systems, ISSR and SCoT, resolved them mostly based on their mixed genomes, especially the triploid ones. In some cases, homo and hetero diploids, triploids and tetraploids clustered together. Both ISSR and SCoT markers distinguished the accessions into three clusters. Existence of homo and hetero accessions was also reported by de Jesus et al. (2013), where major groups (A or AB) regarded as the main clusters correlated with classification of subgroups of ‘Pome’ (including derived hybrids), ‘Silk’, ‘Pisang Awak’, ‘Cavendish’, ‘Gros Michel’, and ‘Plantain’. Although there were some discrepancies, triploid genomes were well resolved by both markers, especially the ones that associated with ISSR. The clustering of the mixed genomes could be due to mislabeling and handling errors as reported (Hippolyte et al. 2012). It could also be linked to inefficiency of the markers to resolve them well based on homo, hetero, diploid, triploid or tetraploid. Population structure of the accessions obtained with ISSR distinctly resolved them into three clusters without any admixture occurring among the accessions and this contradicted the output from SCoT marker system that resulted to admixtures among the accessions identified within the clusters. de Jesus et al. (2013) reported this similar grouping of accession with variable genomes (diploids, triploids, tetraploids) in one cluster or different clusters.
The multilocus structure indices obtained with ISSR and SCoT markers varied across the populations, as suggested by the large portion of the disequilibrium variance (VD) in the total variance from ISSR (VD = 40.94%) and SCoT (VD = 55.03%) in all the accessions. It was also found that Wahlund’s effect (WH) was small in all the populations based on the values from the two marker systems (ISSR, WH = 0.4951; and SCoT, WH = 0.6275), indicating that selection, rather than population admixture or founder effect, was largely responsible for the observed multilocus structure.
Conclusion
Knowledge of distribution of genetic variability, and relatedness among populations or accessions contributes significantly toward crop improvement. The two marker systems demonstrated high effectiveness in resolving GD among the different accessions of Musa species with diverse genomic constitutions. Due to apparent display of higher GD indices (PIC, gene diversity, alleles, Ne, H, I, NPL, PPL, Ht, Hs, GST and Nm) by SCoT, AS genomic group, was considered the most genetically endowed accession. The markers identified higher genetic variability within the populations, with SCoT markers exhibiting higher marker efficiency indicators (PIC, NPL and PPL) in comparison with ISSR. Although the cluster analysis from ISSR and SCoT markers separated them into different groups, the population structure obtained with SCoT provided a more detailed clustering pattern with admixtures in some of the accessions of variable ploidy compositions. Interestingly, SCoT markers, the functional gene-based DNA markers targeting the conserved regions of genome across various plant species, could be more advantageous, informative and efficient in exploration of GD for selection of potential breeding populations of Musa species for improvement and germplasm conservation.
Availability of data and materials
All data generated during this study are included in this published article [and its supplementary information files].
References
Abarshi MM, Mohammed IU, Wasswa P, Hillocks RJ, Holta J, Legg JP, Seal SE, Maruthi MN (2010) Optimization of diagnostic RT-PCR protocols and sampling procedures for the reliable and cost-effective detection of Cassava brown streak virus. J Virol Methods 163:353–359
Andersen JR, Lübberstedt T (2003) Functional markers in plant. Trends Plant Sci 8:554–560
Baurens FC, Martin G, Hervouet C, Salmon F, Yohome D, Ricci S, Rouard M, Habas R, Lemainque A, Yahiaoui N, D’Hont A (2018) Recombination and large structural variations shape interspecific edible bananas genomes. Mol Biol Evol 36(1):97–111
Bhat KV, Jarret RL, Rana RS (1995) DNA profiling of banana and plantain cultivars using random amplified polymorphic DNA (RAPD) and restriction fragment length polymorphism (RFLP) markers. Electrophoresis 16(1):1736–1745
Bhat KV, Amravati Y, Gautam PL, Velayudhan KC (2004) AFLP characterization and genetic diversity analysis of Indian banana and plantain cultivars (Musa spp.). Plant Genetic Res 2(2):121–130
Brown AHD, Feldman MW (1981) Population structure of multilocus associations. Proc Natl Acad Sci USA 78:5913–5915
Cao PJ, Yao QF, Ding BY et al (2006) Genetic diversity of Sinojackia dolichocarpa (Styracaceae), a species endangered and endemic to China, detected by inter-simple sequence repeat (ISSR). Biochem Syst Ecol 34:231–239
Chab MC, Dassou AG, Dossou-Aminon I, Ogouchoro D, Aman BO, Dansi A (2018) Banana and plantain production systems in Benin: ethnobotanical investigation, varietal diversity, pests, and implications for better production. J Ethnobiol Ethnomed 14:78
Christelova P, De Langhe E, Hribova E, Cızkova J, Sardos J, Husakova M, Van den Houwe I, Sutanto A, Kepler AK, Swennen R, Roux N, Dolezel J (2017) Molecular and cytological characterization of the global Musa germplasm collection provides insights into the treasure of banana diversity. Biodivers Conserv 26:801–824
Collard BCY, Mackill DJ (2009) Start codon targeted (SCoT) polymorphism: a simple, novel DNA marker technique for generating gene targeted markers in plants. Plant Mol Biol Rep 27:86–93
Davey M (2013) A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter-and intra-specific Musa hybrids. BMC Genom 14:683
de Jesus ON, de Silva S, Amorim EP, Ferreira CF, de Campos JS, Silva GG, Figueira A (2013) Genetic diversity and population structure of Musa accessions in ex situ conservation. BMC Plant Biol 13:41
Dempewolf H, Eastwood RJ, Guarino L, Khoury CK, Müller JV, Toll J (2014) Adapting agriculture to climate change: a global initiative to collect, conserve, and use crop wild relatives. Agroecol Sustain Food Syst 38:369–377
Doebley JF, Gaut BS, Smith BD (2006) The molecular genetics of crop domestication. Cell 127:1309–1321
Ehrlicha PR, Harte J (2015) Opinion: to feed the world in 2050 will require a global revolution. PNAS 112:14743–14744
Etminan A, Pour-Aboughadareh A, Mohammadi R, Ahmadi-Rad A, Noori A, Mahdavian Z, Moradi Z (2016) Applicability of start codon targeted (SCoT) and inter-simple sequence repeat (ISSR) markers for genetic diversity analysis in durum wheat genotypes. Biotechnol Biotec Equip 30:1075–1081
FAO (Food and Agriculture Organization) (2017) The future of food and agriculture—trends and challenges. Rome 2017
FAO (2019) Food and Agriculture Organization. 2019 edition of the FAO Food Outlook, pp 13–20. http://www.fao.org/3/CA6911EN/CA6911EN.pdf
FAOStat (2014) FAO production statistics for banana and plantain 2012. http://faostat.fao.org/site/567/DesktopDefault.aspx?PageID¼567#ancor. Accessed 20 Mar 14
Ford-Lloyd BV, Schmidt M, Armstrong SJ, Barazani O, Engels J, Hadas R, Hammer K, Kell SP, Kang D, Khoshbakht K, Li Y, Long C, Lu B, Ma K, Nguyen VT, Qiu L, Ge S, Wei W, Zhang Z, Maxted N (2011) Crop wild relatives-undervalued, underutilized and under threat? Bioscience 61(7):559–565
Galvan MZ, Bornet B, Balatti PA et al (2003) Inter simple sequence repeat (ISSR) marker as a tool for the assessment of both genetic diversity and gene pool origin in common bean (Phaseolus vulgaris L.). Euphytica 132:297–301
Ge XJ, Liu MH, Wang WK, Schaal BA, Chiang TY (2005) Population structure of wild bananas, Musa balbisiana, in China determined by SSR fingerprinting and cpDNA-RFLP. Mol Ecol 14:933–944
Godwin ID, Aitken EAB, Smith LW (1997) Application of inter-simple sequence repeat (ISSR) markers to plant genetics. Electrophoresis 18(9):1524–1528
Guo DL, Zhang JY, Liu CH (2012) Genetic diversity in some grape varieties revealed by SCoT analyses. Mol Biol Rep 39:5307–5313
Hajjar R, Hodgkin T (2007) The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica 156:1–13
Hamidi H, Talebi R, Keshavarzi F (2014) Comparative efficiency of functional gene-based markers, start codon targeted polymorphism (SCoT) and conserved DNA-derived polymorphism (CDDP) with ISSR markers for diagnostic fingerprinting in wheat (Triticum aestivum L.). Cereal Res Commun 42(4):558–567
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B Biol Sci 351:1291–1298
Hippolyte I, Jenny C, Gardes L, Bakry F, Rivallan R, Pomies V, Cubry P, Tomekpe K, Risterucci AM, Roux N, Rouard M, Arnaud E, Kolesnikova-Allen M, Perrier X (2012) Foundation characteristics of edible Musa triploids revealed from allelic distribution of SSR markers. Ann Bot 109:937–951
Hogbin PM, Peakall R (1999) Evaluation of the contribution of genetic research to the management of the endangered plant Zieria prostrata. Conserv Biol 13:514–522
Hu W, Zuo J, Hou X, Yan Y, Wei Y, Liu J et al (2015) The auxin response factor gene family in banana: genome-wide identification and expression analyses during development, ripening, and abiotic stress. Front Plant Sci 6:742. https://doi.org/10.3389/fpls.2015.00742
IBA (2007) International Banana Association Banana facts: nutrition. http://www.eatmorebananas.com/facts/nutrition.htm. Accessed 18 Apr 2007
Igwe DO, Afiukwa CA, Ubi BE, Ogbu KI, Ojuederie OB, Ude GN (2017) Assessment of genetic diversity in Vigna unguiculata L. (Walp) accessions using inter-simple sequence repeat (ISSR) and start codon targeted (SCoT) polymorphic markers. BMC Genet 18:98
Ilves A, Lanno K, Sammul M, Tali K (2013) Genetic variability, population size and reproduction potential in Ligularia sibirica (L.) populations in Estonia. Conserv Gen 14:661–669
INIBAP (2008) International network for the improvement of banana and plantain of banana-food and wealth, http://www.inibap.org
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405
Jombart T, Collins C (2015) A tutorial for discriminant analysis of principal components (DAPC) using adegenet 2.0.0. Imperial College London, MRC Centre for Outbreak Analysis and Modelling, pp 1–43
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Karamura E, Frison E, Karamura DA, Sharrock S (1998) Banana production systems in eastern and southern Africa. In: Picq C, Foure E, Frison E (eds) Bananas and food security. INIBAP, Montpellier, pp 401–412
Kastner T, Rivas MJI, Koch W, Nonhebel S (2012) Global changes in diets and the consequences for land requirements for food. Proc Natl Acad Sci USA 109(18):6868–6872
Khayat E, Ortiz R (2011) Genetics of important traits. In: Pillay M, Tenkouano A (eds) Banana breeding. Progress and challenges. CRC Press, Boca Raton, pp 71–84
Khoury CK, Bjorkman AD, Dempewolf H, Ramirez-Villegas J, Guarino L, Jarvis A, Rieseber LH, Struik PC (2014) Increasing homogeneity in global food supplies and the implications for food security. PNAS 111:4001–4006
Kumar A, Mishra P, Singh SC, Sundaresan V (2014a) Efficiency of ISSR and RAPD markers in genetic divergence analysis and conservation management of Justicia adhatoda L., a medicinal plant. Plant Syst Evol 300:1409–1420
Kumar PL, Selvarajan R, Iskra-Caruana ML, Chabannes M, Hanna R (2014b) Biology, etiology, and control of virus diseases of banana and plantain. Adv Virus Res 65:229–269
Lamare A, Rao SR (2015) Efficacy of RAPD, ISSR and DAMD markers in assessment of genetic variability and population structure of wild Musa acuminata colla. Physiol Mol Biol Plants 21(3):349–358
Luz GA, Gomes SO, Araujo Neto RB, Nascimento MSCB et al (2015) Molecular characterization of accessions of Cratylia argentea (Camaratuba) using ISSR markers. Genet Mol Res 14:15242–15248
Martínez LE, Cavagnaro PF, Masuelli RW, Zuňiga M (2006) SSR based assessment of genetic diversity in South American Vitis vinifera varieties. Plant Sci 170:1036–1044
Mukunthakumar S, Padmesh P, Vineesh PS, Skaria R, Kumar KH, Krishnan PN (2013) Genetic diversity and differentiation analysis among wild antecedents of banana (Musa acuminata Colla) using RAPD markers. Indian J Biotechnol 12:493–498
Ortiz R (2000) Understanding the Musa genome: an update. Acta Hortic 540:157–168
Padmesh P, Mukunthakumar S, Vineesh PS, Skaria R, Kumar KH, Krishnan PN (2012) Exploring wild genetic resources of Musa acuminata Colla distributed in the humid forests of southern Western Ghats of peninsular India using ISSR markers. Plant Cell Rep 31:1591–1601
Pakseresht F, Talebi R, Karami E (2013) Comparative assessment of ISSR, DAMD and SCoT markers for evaluation of genetic diversity and conservation of landrace chickpea (Cicer arietinum L) genotypes collected from north-west of Iran. Physiol Mol Biol Plants 19(4):563–574
Perrier X, De Langhe E, Donohue M, Lentfer C, Vrydaghs L, Bakry F, Carreel F, Hippolyte I, Horry JP, Jenny C, Lebot V, Risterucci AM, Tomekpe K, Doutrelepont H, Ball T, Manwaring J, De Maret P, Denham T (2011) Multidisciplinary perspectives on banana (Musa spp.) domestication. PNAS 108:11311–11318
Pollefeys P, Sharrock S, Arnaud E (2004) Preliminary analysis of the literature on the distribution of wild Musa species using MGIS and DIVA-GIS Montpellier, France, INIBAP
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Raboin LM, Carreel F, Noyer JL, Baurens FC, Horry JP, Bakry F, Du Montcel HT, Ganry J, Lanaud C, Lagoda PJL (2005) Diploid ancestors of triploid export banana cultivars: molecular identification of 2n restitution gamete donors and n gamete donors. Mol Breed 16:333–341
Rohlf FJ (2000) NTSYS-pc: numerical taxonomy and multivariate analysis system, version 2.2. Exeter Software. Setauket, New York.
Rotchanapreeda T, Wongniam S, Swangpol SC, Chareonsap PP, Sukkaewmanee N, Somana J (2015) Development of SSR markers from Musa balbisiana for genetic diversity analysis among Thai bananas. Plant Syst Evol. https://doi.org/10.1007/s00606-015-1274-2
Sardos J, Christelová P, Čížková J, Paofa J, Sachter-Smith GL, Janssens SB, Rauka G, Ruas M, Daniells JW, Doležel J, Roux N (2018) Collection of new diversity of wild and cultivated bananas (Musa spp.) in the Autonomous Region of Bougainville, Papua New Guinea. Genet Resour Crop Evol Online first: 25th September 2018. ISSN: 0925–9864.
Scariot V, de Keyser E, Handa T, de Riek J (2007) Comparative study of the discriminating capacity and effectiveness of AFLP, STMS and EST markers in assessing genetic relationships among evergreen azaleas. Plant Breed 126(2):207–212
Silva AVC, Nascimento ALS, Vitória MF, Rabbani ARC, Soares ANR, Lédo AS (2016) Diversity and genetic stability in banana genotypes in a breeding program using inter simple sequence repeats (ISSR) markers. Genet Mol Res 16(1):16019402
Simmonds NW (1966) Bananas, 2nd edn. Longmans, London, p 512
Simmonds NW, Shepherd K (1995) Taxonomy and origins of cultivated Bananas. J Linn Soc Lond, Bot 55:302–312
Souframanien J, Gopalakrishna T (2004) A comparative analysis of genetic diversity in blackgram genotypes using RAPD and ISSR markers. Theory Appl Gen 109:1687–1693
Tang H, Sezen U, Paterson AH (2010) Domestication and plant genomes. Curr Opin Plant Biol 13:160–166
Tripathi L, Odipio J, Tripathi JN, Tusiime G (2007) A rapid technique for screening banana cultivars for resistance to Xanthomonas wilt. Eur J Plant Pathol 121:9–19. https://doi.org/10.1007/s10658-007-9235-4
Ude G, Pillay M, Nwakanma D, Tenkouano A (2002) Analysis of genetic diversity and sectional relationships in Musa using AFLP markers. Theor Appl Genet 104:1239–1245
Ude G, Dimpka CO, Anegbeh PO, Saibu AA, Tenkouano A, Pillay M, Tchoundjeu Z (2003a) Genetic diversity in an African plantain core collection using AFLP and RAPD markers. Theor Appl Genet 107:248–255
Ude G, Pillay M, Ogundwin E, Tenkouano A (2003b) Genetic diversity in an African plantain core collection using AFLP and RAPD markers. Theor Appl Genet 107:248–255
Van Zonneveld M, Scheldeman X, Escribano P, Viruel MA, van Damme P, Garcia W (2012) Mapping genetic diversity of cherimoya (Annona cherimola mill.) and application of spatial analysis of conservation and use of plant genetic. PLoS ONE 7(1):e29845
Vanhove AC, Vermaelen W, Panis B, Swennen R, Carpentier SC (2012) Screening the banana biodiversity for drought tolerance: can an in vitro growth model and proteomics be used as a tool to discover tolerant varieties and understand homeostasis. Front Plant Sci 3:176. https://doi.org/10.3389/fpls.2012.00176
Vinceti B, Loo J, Gaisberger H, Van Zonneveld MJ, Schueler S, Konrad H (2013) Conservation priorities for Prunus africana defined with the aid of spatial analysis of genetic data and climatic variables. PLoS ONE 8(3):e59987
Voisin M, Engel CR, Viard F (2005) Differential shuffling of native genetic diversity across introduced regions in a brown alga: aquaculture vs. maritime traffic effects. Proc Natl Acad Sci USA 102:5432
Yeh FC, Boyle TJB (1997) Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg J Bot 129:157
Ying L, Xin Z, Jinji P, Yanxian Q et al (2011) Molecular assessment of genetic identity and genetic stability in banana cultivars (Musa spp.) from China using ISSR markers. Aust J Crop Sci 5:25–31
Youssef M, James AC, Rivera-Madrid R, Ortiz R, Escobedo-Graciamedrano RM (2011) Musa genetic diversity revealed by SRAP and AFLP. Mol Biotechnol 47:189–199
Zawko G, Krauss SL, Dixon KW, Sivasithamparam K (2001) Conservation genetics of the rare and endangered Leucopogon obtectus (Ericaceae). Mol Ecol 10:2389–2396
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeats (SSR)-anchored polymerase chain reaction amplification. Genom 20:176–183
Funding
This research was supported by National Science Foundation (NSF) with research grant (with Grant Number HRD-1438902) to conduct this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
This work does not involve living animals and no consent is needed.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Igwe, D.O., Ihearahu, O.C., Osano, A.A. et al. Assessment of genetic diversity of Musa species accessions with variable genomes using ISSR and SCoT markers. Genet Resour Crop Evol 69, 49–70 (2022). https://doi.org/10.1007/s10722-021-01202-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-021-01202-8