
Abstract
The 1970s and 1980s saw the appearance of many papers on the topics of synergy, antagonism, and similar concepts of causal interactions and interdependence of effects, with a special emphasis on distinguishing these concepts from that of statistical interaction – the need for a product term in a model. As an example, Miettinen defined “synergism” as “the existence of instances in which both risk factors are needed for the effect”, whereas “antagonism” is where “at least one [factor] can block the solo effect of the other”. In response, Greenland and Poole constructed a systematic analysis of 16 possible individual response patterns in a deterministic causal model for two binary exposure variables, and showed how these patterns can be mapped onto nine types of sufficient causes, which in turn can be simplified into four intuitive categories. Although these and other papers recognized that epidemiology cannot directly study biological mechanisms underlying interaction, they showed how it can usefully study causal and preventive interdependence – which, despite its mechanistic agnosticism, has important implications for clinical decision making as well as for public health.
Similar content being viewed by others


Introduction
In 1980 two papers appeared on interaction and effect modification [1, 2] commenting on debates occurring in the 1970s [3,4,5,6,7]. These papers distinguished between different concepts of interaction: statistical interaction, biologic interaction, public-health interaction, and interaction in individual decision-making (including clinical decision making[8]). Statistical interaction involves a departure from the additivity assumptions in a generalized-linear regression model (usually of a linear, loglinear or logistic form), as represented by the need for a product term in the model. Nonetheless, these papers argued that interaction in public health or in individual decision-making involves a departure from simple additivity of causal risk differences, insofar as the costs of these decisions corresponded to the number of cases generated or prevented by each combination of the exposures [1, 3, 9].
Table 1 gives an example of the type of situation that sparked these debates. It shows the risk of lung cancer in people having two different exposures: E1 and E2, where we assume there is no confounding; for concreteness we may suppose that E1 = asbestos and E2 = smoking. The risk of lung cancer with both exposures (35/1000) is greater than would be expected from adding the separate effects of E2 in the absence of E1 (9/1000), and E1 in the absence of E2 (4/1000) above the background risk (1/1000), which would be 1/1000 + 9/1000 + 4/1000 = 14/1000. The excess over additivity, here 35/1000 − 14/1000 = 21/1000, had been taken as a measure of “synergy” or “biologic interaction” [1, 5, 6], but that usage left some ambiguity in the meaning of those terms. Rothman et al[1] defined biologic interaction as “the interdependent operation of two or more causes to produce disease”. However, “interdependent operation” was not defined in biological terms, and other authors [2, 10] raised objections based on the multistage model of carcinogenesis[11], noting that “the rather general and simple structure of the additive and multiplicative models, as currently used, seems to militate against their ability to reflect closely the sequence of specific and varied biologic events which manifest macroscopically in the form of different diseases”[2].
The situation was made more confusing by differences in the usage of related terms such as “synergy”, and the concurrent use of the related but different term “effect modification”. Thus, it is not surprising that later commentators questioned whether we should use terms like “biological interaction” at all[12, 13]. Miettinen [14] instead shifted the discussion to a framework that left mechanistic elements unspecified. The term “biologic” doesn’t appear until the final discussion, and the focus is instead on “interdependence”. In this context “synergism” is defined as “the existence of instances in which both risk factors are needed for the effect”. This definition is in line with Hume’s definition of causation (often regarded as the founding concept for counterfactuals or potential outcomes)[15, 16] where a factor can be considered a cause when “if the first object had not been, the second never had existed.”[17] In this context, we can consider the combination of two factors to be a cause of disease, if at least some cases of the disease would not have occurred if either factor had been missing. Miettinen then defined antagonism to mean that “at least one [factor] can block the solo effect of the other”. This definition of antagonism is intuitive, but introduces a subtlety that Miettinen missed: in at least one basic interpretation, competitive action becomes a form of antagonism [18].
Simple interdependence
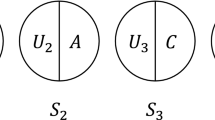
Let us return to Table 1, this time assuming that the effects seen there are only causal (never preventive), and in a sense to be made precise there is only potential synergy (no antagonism) between their effects. This situation is illustrated in Fig. 1, which shows the potential outcomes for the hypothetical cohort of 1,000 people in Table 1, framed in terms of Rothman’s sufficient-component cause model [19]. Removing smoking alone would prevent not only the “jointly caused cases” (21/35), but also the cases due to smoking (but not asbestos) (9/35), i.e. 30/35 = 86% of cases. Similarly, removing asbestos exposure alone would prevent 25/35 = 61% of cases. These two single-factor causal risk differences sum to 147%, which is logically impossible for risk differences. Thus, this sum cannot be the risk difference for the combined effect of smoking and asbestos relative to no exposure.[19] This non-additivity arises because 21 of the 35 cases occurred only because of the joint action of the two exposures.

Numbers of cases occurring through background factors, exposure A alone, exposure B alone, or their combination
This example tells us that some cases occur due to the combination of asbestos exposure and smoking exposure, but it doesn’t tell us how this happens in any mechanical sense. It is quite possible that smoking acts at an early stage in the carcinogenic process (sometimes termed “initiation”), whereas asbestos acts at a later stage (sometimes termed “promotion”)[2]. If so, hypothetically, someone could develop lung cancer because they smoked at age 18–20, and then worked with asbestos at age 45–65. The two exposures need not even be in the body at the same time, and it therefore might seem odd to regard this as a biological interaction if the latter is thought of as a physiological action requiring simultaneity of the exposures. Nonetheless, if some cases occur because the effects of a later exposure depend on the effects of earlier exposure, then we would say there is causal interdependence (or synergy) between the effects of the two exposures.
This “synergism” tells us little about the underlying biologic mechanisms, because many different biological models could produce the same risk patterns[10, 13, 18, 20]. For example, multistage models of cancer[11] hypothesize that carcinogenesis is a multistep process, with the steps being distinct and occurring in a particular order. Such models can predict well the age patterns of epithelial-cancer incidence when that incidence is low, and have more recently been found to yield reasonable predictions for motor neurone disease[21] and for the case fatality of Covid-19[22]. Nonetheless, one can construct two-stage models that reproduce observed incidence patterns to the same degree of accuracy as models with more steps [23].
Individual susceptibilities
Epidemiology is ultimately about populations rather than individuals; that is why we focus on population measures such as average risks (proportions falling ill, or more generally distributions of incidence across time), which are almost always greater than 0 and less than 1. As did Rothman [19], however, Miettinen started from a deterministic concept of individual susceptibility, in which some individuals who have the other relevant (if mostly unmeasured) exposures or cofactors will always develop the outcome if they are exposed to E1, whereas others who do not have enough relevant cofactors will not develop the outcome. He then discussed the implications of susceptibility distributions for population risk patterns. In this model, individuals have an all-or-nothing outcome, coded as 1 or 0. What we see in a population are averages over these all-or-nothing outcomes, which we call incidence proportions (as seen in Table 1). Miettinen gave examples to show how the simple causal model that we assumed above is only one of several that can account for observed risk patterns, given that “correlatedness of susceptibilities” [24] may vary.
To see this identification problem exists even when the exposures are solely causal (never preventive), suppose we have a cohort of just two participants, and that for participant 1 either exposure E1 or E2 or both will produce the outcome (a “competitive-action” response type [18]), whereas for participant 2 only E1 + E2 together will produce it (a “synergistic-action” type). Then, at the level of the cohort of the two participants, the effects on average risks (incidence proportions) of either E1 alone or E2 alone relative to no exposure (the causal risk differences) are (1 + 0)/2 = (0 + 1)/2 = ½ = 50%, while the effect of joint (combined) exposure E1 + E2 relative to no exposure is (1 + 1)/2 = 1 = 100%. This pattern might naively be taken to indicate an absence of synergistic responders in the cohort (because the joint effect is the sum of the single effects), whereas in fact the response pattern of participant 2 exhibits synergism (the outcome only occurs because of the combination of exposures E1 and E2). Exactly the same pattern would however arise if participant 1 got the outcome exactly and only when E1 was present, and participant 2 got the outcome exactly and only when E2 was present, so that there was no synergism at all. Thus, a given incidence pattern in a cohort can be consistent with very different patterns of exposure susceptibilities.
Greenland and Poole [18] responded to Miettinen’s paper with a systematic analysis of a table of the 24 = 16 possible individual response patterns allowed by Miettinen’s deterministic causal model with two binary exposures (and thus 4 possible exposure combinations, as in Table 1) and a binary outcome, allowing that a particular exposure can cause or prevent disease depending on the other factor. Contrary to Miettinen, they pointed out that, in this model, causation and prevention could be treated as symmetric properties. A consequence is that additivity could be derived from a no-interdependence assumption, provided the competitive-action response type (someone who gets the outcome if exposed to E1, E2, or both) was treated as a form of antagonism or effect interference, and thus is ruled out by a no-interdependence assumption [18]. For such a type, if E1 comes first then the outcome will occur regardless of E2, and thus E1 is blocking any and all further action by E2 on the outcome; if E2 comes first then then the outcome will occur regardless of E1 and thus E2 is blocking half of any further action by E1. Hence, in either case we can say competitive response types exhibit antagonism in that one factor blocks the effect of the other.
Greenland and Poole also showed how the 16 deterministic types can be mapped onto 9 types of sufficient causes, showing again the symmetries of causation and prevention, which in turn can be simplified into four intuitive categories illustrated by the causal pies in Fig. 1. There, we let factors such as “E1” in Fig. 1 stand for both the case where E1 is causal (E1 = 1 in the figure) and the case where E1 is preventive (E1 = 0, i.e., the absence of E1 is necessary for this particular sufficient cause). In this situation, the four invariant properties (under recoding) are no involvement of either variable (first sufficient cause in Fig. 1), involvement of only E1 (2nd sufficient cause), involvement of only E2 (3rd ), or involvement of both (4th ).
A more extensive review of the background theory can be found in Greenland et al. [25].
Further history
Although Miettinen [14] provided a thoughtful conceptual analysis, the models and mathematical results in the paper were not new. For example, there were already many articles with individual causal models going back to at least the 1920s. Neyman [26] is often cited as the first such paper, and by the 1960s the bioassay literature was using what Miettinen called causally interdependent individual response types to derive response probabilities; see Ashford and Cobby [27] and Weinberg [28] for overviews of that early literature.
By the mid-1970s, epidemiological papers also began deriving population risk patterns from individual models of synergism and other interdependencies. In particular, Rothman in 1976 [19] and Koopman in 1981 [29] had done so using a sufficient-component cause model, while Hamilton in 1979 [30] and Wahrendorf and Brown in 1980 [24] had used instead a potential-outcome model like Neyman’s. Thus, although the concept of building population risks from individual susceptibilities was an old one, a series of papers starting in the mid-1970s introduced the idea to the ongoing epidemiological discussions about interaction and interdependence in a clear and coherent manner, and these seminal papers continue to influence current thinking on causality[31, 32]. In addition, a number of useful results for detecting interdependent effects via departures from risk additivity have been verified and derived [32,33,34,35,36,37].
An objection to the above approaches is the lack of statistical power at typical sample sizes for detecting departures from additivity[38]. Another is that they are based on deterministic causal models. To address that objection, Greenland and Poole [18] also described a model of independent action based on stochastic potential outcomes [33, 37, 39] which leads to additive hazard-rate differences when there is no interdependence of effects. The latter additivity also follows from the model in Wahrendorf and Brown [24] and Weinberg [28], which as they discussed was derived from much earlier bioassay literature on independent action.
Discussion
Given the many factors that influence population data, one might question how useful these conceptual modelling exercises are for epidemiology. For most of the models we have found that it is possible to describe realistic scenarios in which they at least provide useful thought experiments to test speculative hypotheses. For example, it is easy to find examples in which different medications given separately may prevent adverse events, albeit not necessarily in the same patients, but may be antagonistic in the sense that their combination actually increases the risks in some patients (because of drug synergies) – a major concern in the topic of polypharmacy. On the other hand, one can conceive of examples where both factors may increase the risk of an adverse outcome, but their combination may or may not be mutually antagonistic and prevent the outcome depending on the specifics of the mechanism. For example, Miettinen noted that drugs which can cause severe acidosis or alkalosis may kill some patients, but careful combination might be harmless [14]. The timing and other specifics of drug combination are beyond the crude labelling typical in basic “interaction” models seen in epidemiology and biostatistics, although they are of clear concern in the bioassay and pharmacology literatures.
Regardless of whether the models are realistic, in light of the earlier literature [27] one cannot justify Miettinen’s claim that “until now the conceptualization of synergism has been burdened by epistemological and statistical concerns, whereas in this paper… the take-off is guided by ontological considerations alone”[14]. A particular problem for Miettinen’s approach is that its definition of independent action depends on the choice of reference categories for the exposures. As Greenland and Poole noted [18], Miettinen constructed his definition on the assumption that causal and preventive action could be sharply distinguished as ontological concepts. This can be the case in situations in which the sequence of actions is clear, as with viral infections and vaccines. But often causal and preventive actions are distinguished only by arbitrary coding and thus there is no “correct” reference group (e.g. when considering gender or ethnicity).[18].
What is the legacy of the numerous papers on synergy, antagonism, and causal interaction that appeared in the 1970 and 1980 s? We think they laid out a number of scenarios and issues which remain useful and relevant. For example, the use of individual susceptibilities to derive population risks and rates has become mainstream, as seen in several current textbooks[31, 32]; the book by VanderWeele [32] in particular goes into great depth about modelling interdependent effects. On the other hand, as many have acknowledged, rarely can we identify hypothesized deterministic individual susceptibilities, so the approach may be of little use for risk prediction or clinical practice. To address this problem, subsequent authors borrowed the concept of stochastic (random) individual potential outcomes from the bioassay literature [18, 31, 37], noting in particular how certain common interpretations of measures of causal attribution corresponded to a (usually implausible) assumption of “no biologic interaction with background factors”.[33].
Conclusion
Epidemiologists and statisticians analyse groups, not individuals. Suppose (even if only as a thought experiment) we are prepared to make a number of assumptions, including that: (a) susceptibilities and responses are uncorrelated across individuals and follow a simple potential-outcomes (counterfactual) model; (b) the response types do not counterbalance one another to produce a causally “unfaithful” risk pattern; and of course (c) there is no uncontrolled bias. In this situation, we can follow the relatively straightforward approach summarized above in Table 1 and Fig. 1, in which we say causal interdependence is present when at least some effects of one of the exposures occur or are prevented because of the other exposure. When that occurs, the joint effect of the two exposures will be more than additive when synergistic responders outnumber antagonistic responders, and less than additive when the opposite is the case. In this regard it is essential that, when such issues are being considered, the separate and joint effects of exposures are presented (as in Table 1) rather than dealt with simply by adding product (“interaction”) terms to regression models.
Nonetheless, as discussed above, epidemiologic data alone can tell us little about the biological mechanisms underlying risk patterns, which is why some authors[12, 13] argued that the term “biological interaction” is best avoided in epidemiology (even if it remains useful in physiological studies). This problem may be seen in that the same pattern of interdependence can arise from different time orderings of the exposures, and thus from what may be very different mechanisms of action. By definition, epidemiology does not study such mechanisms directly, but it can usefully study causal interdependence – which, despite its mechanistic agnosticism, has important implications for clinical decision making as well as for public health.
References
Rothman KJ, Greenland S, Walker AM. Concepts of interaction. Am J Epidemiol. 1980;112(4):467–70.
Saracci R. Interaction and synergism. Am J Epidemiol. 1980;112(4):465–6.
Blot WJ, Day NE. Synergism and interaction: are they equivalent? Am J Epidemiol. 1979;110(1):99–100.
Kupper LL, Hogan MD. Interaction in epidemiologic studies. Am J Epidemiol. 1978;108(6):447–53.
Rothman KJ. Synergy and antagonism in cause-effect relationships. Am J Epidemiol. 1974;99(6):385–8.
Rothman KJ. The estimation of synergy or antagonism. Am J Epidemiol. 1976;103(5):506–11.
Walter SD, Holford TR. Additive, multiplicative, and other models for disease risks. Am J Epidemiol. 1978;108(5):341–6.
Corraini P, Olsen M, Pedersen L, Dekkers OM, Vandenbroucke JP. Effect modification, interaction and mediation: an overview of theoretical insights for clinical investigators. Clin Epidemiol. 2017;9:331–8.
Pearce N. Analytical implications of epidemiological concepts of interaction. Int J Epidemiol. 1989;18(4):976–80.
Siemiatycki J, Thomas DC. Biological models and statistical interactions: an example from multistage carcinogenesis. Int J Epidemiol. 1981;10(4):383–7.
Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8(1):1–12.
Lawlor DA. Biological interaction: time to drop the term? Epidemiology. 2011;22(2):148–50.
Thompson WD. Effect modification and the limits of biological inference from epidemiologic data. J Clin Epidemiol. 1991;44(3):221–32.
Miettinen OS. Causal and preventive interdependence. Elementary principles. Scand J Work Environ Health. 1982;8(3):159–68.
Greenland S, Robins J, Pearl J. Confounding and collapsibility in causal inference. Stat Sci. 1999;14:29–46.
Pearce N, Vandenbroucke JP. Educational note: types of causes. Int J Epidemiol. 2020;49(2):676–85.
Hume D. An Enquiry Concerning Human Understanding. Oxford: Oxford University Press; 2007.
Greenland S, Poole C. Invariants and noninvariants in the concept of interdependent effects. Scand J Work Environ Health. 1988;14(2):125–9.
Rothman KJ, Causes. Am J, Epidemiol. 1976. 104(6): p. 587–92.
VanderWeele TJ. Sufficient cause interactions and statistical interactions. Epidemiology. 2009;20(1):6–13.
Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, Heverin M, Howard RS, Huisman MHB, Keren N, Leigh PN, Mazzini L, Mora G, Orrell RW, Rooney J, Scott KM, Scotton WJ, Seelen M, Shaw CE, Sidle KS, Swingler R, Tsuda M, Veldink JH, Visser AE, van den Berg LH, Pearce N. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13(11):1108–13.
Pearce N, Moirano G, Maule M, Kogevinas M, Rodo X, Lawlor DA, Vandenbroucke J, Vandenbroucke-Grauls C, Polack FP, Custovic A. Does death from Covid-19 arise from a multi-step process? Eur J Epidemiol. 2021;36(1):1–9.
Moolgavkar SH. Model for human carcinogenesis: action of environmental agents. Environ Health Perspect. 1983;50:285–91.
Wahrendorf J, Brown CC. Bootstrapping a basic inequality in the analysis of joint action of two drugs. Biometrics. 1980;36(4):653–7.
Greenland S, Rothman KJ, Lash TL, Concepts of interaction, in Ch. 5 in Modern Epidemiology, 3rd ed. 2008, Lippincott: Philadelphia.
Splawa-Neyman J. On the Application of Probability Theory to Agricultural Experiments. Essay on Principles. Section 9. Stat Sci. 1990;5:465–80.
Ashford JR, Cobby JM. A system of models for the action of drugs applied singly or jointly to biological organisms. Biometrics. 1974;30(1):11–31.
Weinberg CR. Applicability of the simple independent action model to epidemiologic studies involving two factors and a dichotomous outcome. Am J Epidemiol. 1986;123(1):162–73.
Koopman JS. Interaction between discrete causes. Am J Epidemiol. 1981;113(6):716–24.
Hamilton MA. Choosing the parameter for a 2 x 2 table or a 2 x 2 x 2 table analysis. Am J Epidemiol. 1979;109(3):362–75.
Hernan MA, Robins JM. Causal Inference: What If. Boca Raton: Chapman & Hall/CRC; 2020.
VanderWeele T. Explanation in causal inference. New York: Oxford University Press; 2015.
Robins J, Greenland S. The probability of causation under a stochastic model for individual risk. Biometrics, 1989. 45(4): p. 1125-38; Erratum: 1991, 48, 824.
VanderWeele TJ, Knol MJ. Remarks on antagonism. Am J Epidemiol. 2011;173(10):1140–7.
VanderWeele TJ, Robins JM. The identification of synergism in the sufficient-component-cause framework. Epidemiology. 2007;18(3):329–39.
VanderWeele TJ, Robins JM. Empirical and counterfactual conditions for sufficient cause interactions. Biometrika. 2008;95:49–61.
Vanderweele TJ, Robins JM. Stochastic counterfactuals and stochastic sufficient causes. Stat Sin. 2012;22(1):379–92.
Greenland S. Tests for interaction in epidemiologic studies: a review and a study of power. Stat Med. 1983;2(2):243–51.
Greenland S. Interpretation and choice of effect measures in epidemiologic analyses. Am J Epidemiol. 1987;125(5):761–8.
Acknowledgements
We thank Deborah Lawlor, Rodolfo Saracci, Jan Vandenbroucke and Tyler VanderWeele for their comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declarations
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. The authors have no relevant financial or non-financial interests to disclose. NP prepared the initial draft of the paper which was then revised by SG; both authors then contributed to subsequent revisions.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pearce, N., Greenland, S. On the evolution of concepts of causal and preventive interdependence in epidemiology in the late 20th century. Eur J Epidemiol 37, 1149–1154 (2022). https://doi.org/10.1007/s10654-022-00931-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-022-00931-z