
Abstract
The federally endangered Cape Sable seaside sparrow (Ammospiza maritima mirabilis) is endemic to the Greater Everglades ecosystem in southern Florida, inhabiting fragmented marl prairies in six individual subpopulations. The subspecies is threatened by loss of breeding habitat from fire and water management. Genetic information is severely limited for the subspecies but could help inform decisions regarding subpopulation protections and potential translocations for genetic rescue. To provide genetic data and inform management efforts, feather samples were collected across five subpopulations (designated A–E) and protocols were tested to optimize DNA extraction yields. We assessed four mitochondrial DNA markers (N = 36–69) and 12 nuclear microsatellite loci (N = 55) in 108 sparrows. Mitochondrial DNA sequences revealed low haplotype diversity, with NADH dehydrogenase-2 haplotypes matching to most other extant subspecies and to the Atlantic coast subspecies. Nuclear diversity was low compared to other subspecies, but similar across subpopulations. Samples grouped as one population when analyzed by Principal Component Analysis, Bayesian modelling and genetic distance metrics. Limited genetic emigration was detected from one putative migrant. Relatedness was significantly different for sparrows in the most geographically distant subpopulation (A), likely reflecting high self-recruitment and natal site fidelity (P = 0.003). The low to moderate effective population size (NE = 202.4; NE:NC = 0.06) and generation time estimates indicated that unique genetic variation could be lost quickly during stochastic events. The sample sizes were limited, which reduced the power to comprehensively address recent population size reductions and any subsequent loss of genetic diversity.
Similar content being viewed by others


Introduction
The protection of endangered species from population threats, like habitat loss, is needed to effectively manage biological resources. To maintain population viability and evolutionary processes, a number of conservation units are used to delineate and manage demographically independent groups, such as distinct population segments (DPSs) under the United States Endangered Species Act, and evolutionary distinct units (ESUs). Measurement of intraspecific genetic diversity, localized adaptation, and genetic or demographic resilience can be used to inform conservation efforts for imperiled or declining populations (Flanagan et al. 2018; Ottewell and Byrne 2022; Thompson et al. 2023). Mitochondrial and nuclear genetic studies can provide information on genetic variation, connectivity, population structure, relatedness, and inbreeding levels to inform conservation efforts.
The seaside sparrow (Ammospiza maritima) species complex is composed of seven subspecies found in marshes along the Atlantic and Gulf of Mexico coastlines of the United States (McDonald 1988). These seven subspecies were designated by feather plumage, song, and in some instances, mitochondrial DNA (mtDNA), but past and current taxonomic classifications and differentiations are still being finalized by comprehensive genetic and phenotypic analyses (Griscom 1944; Kale 1983; Robbins 1983; McDonald 1988; Mila et al. 2007; Remsen 2010; Winker 2010; Woltmann et al. 2014; Davis et al. 2021; Enloe et al. 2021; Ryan and Woltmann 2023). Within the species complex, only two of the seven subspecies were previously considered their own species and inhabit freshwater marshes: the Dusky seaside sparrow (A. m. nigrescens, now considered extinct) and Cape Sable seaside sparrows (A. m. mirabilis, considered federally endangered) (McDonald 1988). However, contaminants, habitat loss and possibly isolation eventually resulted in the extinction of the Dusky seaside sparrow subspecies (Noecker 1998).
The extant federally endangered Cape Sable seaside sparrow (A. m. mirabilis) is found at the southern edge of the species range and is endemic to southern Florida and Everglades National Park (ENP) in Monroe and Miami-Dade counties. To date, migration or interbreeding of the Cape Sable seaside sparrow with other populations has not occurred due to their geographical isolation (Lockwood et al. 2001). Cape Sable seaside sparrows are restricted to fragmented, short-hydroperiod freshwater marl prairies and have a lifespan of about 5 to 6 years (Lockwood et al. 2001). The marl prairie ecosystem health and appropriate hydroperiods are necessary for Cape Sable seaside sparrows to have consistent and productive breeding season conditions (Sah et al. 2011). However, favorable breeding habitat for the Cape Sable seaside sparrow has been historically altered by the reallocation of waterflow, fires, hurricanes and introductions of exotic plant species, leading to inconsistent breeding years and cryptic population dynamics (Roman et al. 1994; Platt et al. 2002; Lockwood et al. 2003). While other seaside sparrow subspecies may more easily adapt and recover from altered landscapes, the Cape Sable seaside sparrow has not shown this pliancy in habitat usage as their habitat has shifted from short- to long-hydroperiods (Curnutt et al. 1998; Lockwood et al. 2001; Sah et al. 2011).
Currently, characteristics of the Cape Sable seaside sparrow subspecies and population dynamics have been estimated by demographic surveys for the six recognized subpopulations and very limited genetic data (Benscoter and Romañach 2022). The six recognized subpopulations were designated for management purposes by habitat and demographic data (Fig. 1a) (U.S. Fish and Wildlife Service 2020; Virzi and Tafoya 2021). Two previous genetic studies using mitochondrial (n = 5 sparrows) and genome-wide nuclear analysis (n = 2 sparrows) found support for the Cape Sable seaside sparrow subspecies as a distinct population segment of the species and the latter observed potentially low genetic diversity (Nelson et al. 2000; Davis et al. 2021). The mitochondrial haplotypes and genome-wide markers grouped the Cape Sable seaside sparrows closely to the Atlantic coast clade of the species, although the Cape Sable subspecies is likely not interbreeding with other subspecies due to the geographic distance (Nelson et al. 2000; Davis et al. 2021). Mark-recapture and demographic survey data have indicated limited dispersal and greatly fluctuating population sizes among the six subpopulations since the 1990s (Dean and Morrison 2001; Boulton et al. 2009; Van Houtan et al. 2010; Gilroy et al. 2012; Virzi and Tafoya 2021; Benscoter and Romañach 2022). The Cape Sable subspecies was delimited through morphometrics, sonography and the geographic isolation of the southern Florida population (McDonald 1988).

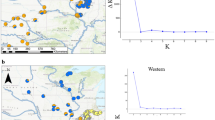
a Geographic distribution of U.S. Fish and Wildlife defined subpopulations, or management units, (A–F) of the Cape Sable seaside sparrow throughout the southern Everglades of Florida, USA near Everglades National Park (ENP) and b haplotype networks for mitochondrial DNA loci (D-loop and ND2) for all management units sampled (A–E). Unit F was not sampled due to logistical challenges. In the haplotype networks, each circle represents a unique haplotype, with the size of the circle relative to the sample size containing that haplotype. The proportional slices are related on the subpopulation where the sparrow was captured. The number of sparrows in each subpopulation with the given haplotype is noted. The hatch marks on the lines connecting the haplotype circles identify the number of mutational differences between respective haplotype sequences. Colors of units A–E match the subsequent figures in this paper
To date, no genetic studies have investigated the patterns of the Cape Sable seaside sparrow subspecies range-wide genetic diversity, historical relationships, or connectivity and relatedness among subpopulations, which can all inform appropriate management strategies (Nelson et al. 2000; Davis et al. 2021; Radford et al. 2021). Cape Sable seaside sparrow subpopulations primary threats are loss of breeding habitat due to high water levels and fire management in the Greater Everglades. Genetic knowledge could help inform decisions regarding sparrow subpopulation protections and the potential for genetic rescue through translocations. Further, the geographical separation of Cape Sable seaside sparrows from other subspecies leads us to hypothesize that the population may have lower genetic diversity levels, similar to previous studies of an isolated southern population of Texas seaside sparrows (A. m. cf. sennetti) (Woltmann et al. 2014, 2019). To inform the management of this endangered and isolated subspecies, this study has two descriptive objectives to: (1) identify and sequence mitochondrial DNA markers to address matrilineal genetic diversity and structure of the recognized subpopulations in Everglades National Park, and (2) classify and multiplex microsatellite markers to address fine-scale, contemporary subpopulation connectivity and relatedness of Cape Sable seaside sparrows.
Study area
Cape Sable seaside sparrows breed in seasonally flooded marl prairie habitat dominated by subtropical graminoids. Demographic studies were conducted annually between April and August of the years 2011 through 2018 by monitoring a total of 3418 hectares of dry suitable breeding habitat. In and around Everglades National Park the suitable habitat areas are referred to as subpopulations A, B, C, D, E and F for management purposes, but genetic data have not confirmed this terminology. Hereafter, ‘subpopulations’ will be referred to as ‘management units’ or ‘units’ to distinguish them from genetically defined subpopulations. Sparrows were sampled in management units A, B, C, D and E (not F due to logistical challenges reaching the site) during the dry breeding season (March-July). Hereafter, ‘all management units’ refers only to units A–E sampled in this study (Fig. 1a). The geographic areas of the management units experience variable water allocations leading to extreme flooding or droughts and changes to vegetation composition from local fire prescriptions (Lockwood et al. 2003). As a result, the management units experience significant local water level differences, including changes in the size of suitable habitat and habitat matrix fluctuations leading to the need for individual management of each unit. The proposed critical habitat areas of the units encompass suitable breeding habitat and range from 71,294 hectares (unit A) to 4958 hectares (unit F) (U.S. Fish and Wildlife Service 2006). The marl prairie habitat vegetation composition fluctuates when areas become wetter or drier due to water reallocation and prescribed burns, thus changing the proportion of potential suitable breeding habitat in each unit (Sah et al. 2021).
Methods
Sample collection
Feather samples were collected from juvenile and adult sparrows between June 2011 and June 2018 and authorized under Federal Bird Banding Permit #23166 and Native Endangered Species Recovery Permit #TE075916 issued by the U.S. Department of the Interior. These permits ensure the study plan was approved by the U.S. Department of the Interior and animals were treated humanely and ethically. Mist nets were utilized to capture Cape Sable seaside sparrows following the protocol described in Bibby et al. (1992). Each sparrow was given a unique identification band at the first capture and each recapture was noted to prevent genetic data from duplicate individuals. Five to eight breast feathers were collected from each bird and all stored dry at room temperature (Taberlet and Luikart 1999).
DNA extraction and amplification
The calamus tip of the small feathers contained minute numbers of cells with limited amounts of DNA. DNA extraction techniques were tested to maximize the quality and quantity of DNA for multi-locus studies and a modified Phenol:Chloroform:Isoamyl Alcohol (PCI) protocol was selected (Sambrook et al. 1989) with further details provided in Online Resource Appendix 1 (Table S1). Batches of five feathers from each sparrow were used in each extraction to increase DNA yield. The modified PCI protocol improved the digestion of keratin in the calamus tips by using Tris-NaCl-EDTA pH 7.5 (1 M Tris-HCl, 5 M NaCl, 0.5 M EDTA) and 1 M Tris-HCl pH 8.0 instead of TE buffer and added proteinase K (20 mg/mL) and 1 M dithiothreitol (DTT) (Sambrook et al. 1989). Glycogen from Blue mussel, Mytilus edulis (1.25 µL/sample; Sigma-Aldrich, St. Louis, Missouri) was added and left overnight at − 20 °C prior to the precipitation of DNA to maximize the chances DNA pellet recovery. DNA quality and quantity was assessed using spectrophotometric absorbance (EPOCH, BioTek Instrument Inc, Winooski, VT) and DNA quality was assessed by purity ratios (260/280).
Mitochondrial DNA (mtDNA) was initially assessed by amplifying 3500 base pairs across five loci: Cytochrome b (Cytb), control region D-loop, NADH dehydrogenase 2 (ND2), 16S ribosomal RNA (16S) and 12S ribosomal RNA (12S), using 14 sets of primers (Table S2) (Nelson et al. 2000; Alström et al. 2008; Ross and Bouzat 2014; Woltmann et al. 2014; Foran et al. 2015). Primers were screened on two Cape Sable seaside sparrow egg samples with high quantities of DNA and optimized on five feather samples for longer fragment lengths, greater nucleotide diversity, and clean amplification. The final set of primers amplified on the sample set were Cytb, D-loop, ND2 and 16S. Mitochondrial amplification reactions were performed in a gradient thermocycler (MJ Research Inc., Watertown, MA). Each 25 µL reaction performed in 96-well microwell plates (Fisher Scientific, Waltham, MA) contained 1 µL of template DNA, 1X PCR Buffer without MgCl2 (Roche, Mannheim, Germany), 2.5 mM MgCl2, 0.5 U Taq polymerase (Sigma, St. Louis, MO), 0.5 µM of each primer, and 0.2 mM of each dNTP. Cycling conditions were as follows: 3 min initial denaturation at 94 °C; 35 cycles, each of 60 s denaturation at 94 °C; 60 s annealing at 57 °C (16S annealing at 49 °C); 60 s extension at 72 °C; and a 10 min final extension at 72 °C. The PCR products were visualized on 2–3.5% (wt/vol) agarose gels in 1X TBE buffer (Tris/Borate/EDTA). All feather samples that amplified successfully were purified by ExoSap-IT (Affymetrix, Santa Clara, CA) and incubated at 37 °C for 30 min then 80 °C for 15 min to inactivate the removal of unused primers and nucleotides. Purified samples were sequenced by Eurofins Genomics (Louisville, KY, USA) on an ABI3700 Sequence Detection System (Applied Biosystems, Foster City, CA).
The sequences were aligned and analyzed using Geneious 10.0.9 (Biomatters, Inc., Auckland, New Zealand, http://www.geneious.com) and Mesquite v 3.51 (Maddison and Maddison 2018). To obtain accurate sequences on the low yield samples, up to three PCR amplifications and sequencing attempts were made. Sequencing was completed in both the forward and reverse directions. The sequence quality was assessed by eye with bases no less than 80% of the expected length retained during trimming. Unique haplotypes were searched using the NCBI BLAST tool to identify matches to previously published haplotypes (Johnson et al. 2008).
Microsatellite loci were identified by testing primers developed in related species and subspecies on the extracted feather samples from 114 sparrows (Bulgin et al. 2003; Hill et al. 2008; Poesel et al. 2009; Sardell et al. 2010; Lehmicke et al. 2012). After identifying suitable amplification success and polymorphism, the 18 loci were compiled by similar annealing temperatures into five different multiplexes (Table S3). Each 10 µL reaction contained 1 µL of template DNA, 1X PCR Buffer without MgCl2 (Roche, Mannheim, Germany), 25 mM MgCl2, 0.5 U Taq polymerase (Sigma, St. Louis, MO), 1 mg/mL Bovine Serum Albumin, 25 µM of each primer, and 0.2 mM of each dNTP. Microsatellite amplification reactions were performed in a gradient thermocycler (MJ Research Inc., Watertown, MA) with the following conditions: 5 min initial denaturation at 95 °C; 34 cycles of 30 s denaturation at 95 °C, 1 min annealing at 62.9 °C for MP4 and MP5 (MP1 at 55 °C, MP2 at 60 °C, MP3 at 61.8 °C) and 1 min extension at 72 °C; and a 10 min final extension at 72 °C. The PCR products were visualized in 1% (wt/vol) agarose gels in 1X TBE buffer (Tris/Borate/EDTA). The positive DNA fragments of all feather samples were run on an ABI3130 xl Genetic Analyzer (Applied Biosystems, Foster City, CA). The alleles were standardized with GeneScan ROX 500 Size Standard (Applied Biosystems, Foster City, CA) and scored using Geneious v. 10.0.9. Each sample and marker combination were analyzed a minimum of three times to ensure genotype accuracy. The same positive control sample was analyzed in triplicate on each plate to assess accuracy across sequencing runs. Each sample was run in triplicate to provide a consensus final genotype call with homozygotes determined when all fragment analyses yielded a homozygote, while heterozygotes were determined when at least two of three analyses yielded a heterozygote. If a sample was not clearly homozygous or heterozygous, three more amplifications were conducted to gain a consensus using the same rules. We calculated observed genotype error rate by subsetting more than 20% of consensus data and recording the number of genotypes erroneously assigned over the total reactions performed. Genotypes were erroneously assigned if the allele calls differed from a consensus genotype in the same plate but were not assessed for samples requiring additional PCRs.
Mitochondrial and microsatellite genetic diversity analyses
Mitochondrial DNA sequence polymorphisms were analyzed using the DNAsp v6.12.03 software (Rozas et al. 2003). Genetic diversity metrics were reported as the number of haplotypes (H), haplotype diversity (h), number of variable sites (S), nucleotide diversity (π), and theta by site and sequence (θ). Each parameter was calculated across all samples for each locus since sample sizes for some management units were low (n ≤ 6). Sequences were not able to be concatenated or assessed as one long fragment since the number of samples sequenced for each locus was highly variable and would have resulted in a reduced number of samples for analysis. Loci with multiple haplotypes were visualized on a simple median joining network (epsilon = 0) in PopART v1.7 software and haplotype frequencies were reported by management unit (Leigh et al. 2015).
All nuclear microsatellite loci were tested for evidence of stuttering errors, null alleles, and short allele dominance in MicroChecker (Van Oosterhout et al. 2004). Departures from expected genotypic frequencies in Hardy–Weinberg equilibrium were evaluated in GenAlEx 6.501 (Peakall and Smouse 2006). Genotypic linkage disequilibrium was evaluated in FSTAT version 2.9.3.2 based on 1320 permutations to test for non-random associations between alleles of different loci (Raymond and Rousset 1995). Alpha values for Hardy–Weinberg equilibrium, linkage disequilibrium and subsequent pairwise comparisons tests were adjusted using the sequential Holm-Bonferroni method, which ranks p-values and adjusts the target alpha based on rank values and the number of total tests to reduce the risk of Type I errors (Holm 1979; Rice 1989). Polymorphism information content (PIC) was estimated in CERVUS 3.0 to provide reference on how informative the loci were when separated into each individual management unit versus one metapopulation and values > 0.5 were considered very informative (Kalinowski et al. 2007).
Genetic diversity was estimated by the average number of alleles per locus (NA), number of private alleles, or alleles unique to a management unit (AP), observed (HO), and expected (HE) heterozygosity, all calculated in GenAlEx 6.501. Allelic richness (AR), or the number of alleles corrected for sample size, and inbreeding coefficients (FIS) were calculated in FSTAT version 2.9.3.2 (Goudet 2002). A randomized block ANOVA test was run in RStudio for each measure of genetic diversity to determine significant differences among management units, using loci as the randomized blocks (R Core Team 2019). If tests found significant differences among the management units, a Dunn’s test was performed to evaluate pairwise comparisons using the R package ‘dunn.test’ (Dinno 2017; R Core Team 2019). The alpha value for multiple comparisons was adjusted using the sequential Holm-Bonferroni method (Holm 1979; Rice 1989). Genetic diversity parameters, NA, AR, HO and HE, were compared to microsatellite results from previous studies of other subspecies to allow for subspecific comparison of genetic diversity. However, given the low sample sizes in the present study, the differential markers used among the studies, and the discordance found between genetic data and current subspecies delineations, these values are not to be used for taxonomic descriptions (Woltmann et al. 2014; Roeder et al. 2021). Although the microsatellite panels across the various subspecies were slightly to moderately different, the subspecies comparison was helpful to provide context for the genetic diversity found within Cape Sable seaside sparrows (concept reviewed in Leigh et al. (2021)). Tests were run in Bottleneck software with 10,000 iterations, a variance of 12 and the two-phase model (TPM) adjusted to 95% stepwise mutation model (SMM) as suggested for microsatellites (Piry et al. 1999). A bottleneck was considered plausible in a management unit if heterozygote excess was significant in TPM or SMM for the Wilcoxon test and a mutation-drift equilibrium was shifted under the allele frequency distribution test.
The generation length and the ratio of effective population size (NE) to total census population size (NC; NE:NC), can both be proxy estimators for how resilient populations are to stochastic events in the environment (Bird et al. 2020; Hoban et al. 2020). We estimated generation length by first calculating the age at first breeding for Cape Sable seaside sparrows using mark-recapture data recorded annually. As detailed in Virzi and Tafoya (2021), adults and juvenile sparrows were leg-banded annually on all demographic study plots, and color-band resighting surveys were conducted every breeding season daily for at least 3 months over a 10 year period (2010–2019). The average age of first breeding was calculated by determining the number of days between each sparrow’s initial banding date as nestlings and the date of first known breeding (Table S4). We followed methodologies and equations provided in Bird et al. (2020) using age at first breeding (F), survival rate (S), and longevity (L) of Cape Sable seaside sparrows to estimate generation length for the subspecies. We calculated both the GFLS and GFL estimates of generation length, using an average value of z dependent on F, and reported the greatest value as generation length (Eq. 1 and 2 in Bird et al. (2020)).
To estimate the rate of genetic loss at the time of sample collection, we started by calculating the contemporary estimates of NE, or the number of sparrows contributing unique diversity to the next generation, considering all management units as one metapopulation in NeEstimator v.1.3 using the linkage disequilibrium method (Peel et al. 2004). The estimated NE was compared to the estimated NC reported from annual range-wide survey data in 2018 to determine the approximate observed NE:NC ratio, which is considered inversely proportional to the rate of genetic erosion and related to the genetic health of a population (Frankham 1995; U.S. Fish and Wildlife Service 2019, 2020; Hoban et al. 2020). The methodology to estimate total population size is outlined in U.S. Fish and Wildlife Service (2020) but briefly, the NC estimate was calculated based on point counts from range wide helicopter surveys, where the number of observed sparrows was corrected as done in Bass and Kushlan (1982) and assuming every male is accompanied by a female (Pimm et al. 2002).
Management unit connectivity analyses
Divergence and connectivity among management units were evaluated using various Bayesian, multivariate, and genetic distance analyses. The admixture model of the Bayesian program Structure was run specifying a burn-in period of 50,000 iterations and a total chain length of 100,000 iterations. Twenty replicate Markov chain Monte Carlos (MCMCs) were examined for each inferred number of populations (K) between 1 and 11 (Falush et al. 2003). The best-supported hypothesis was considered as the K cluster with the highest LnP(K) and the highest ΔK values (Evanno et al. 2005; Earl and von Holdt 2012). A pairwise genetic distance matrix based on allele data among sparrows was calculated in GenAlEx 6.501 and used to plot a principal component analysis (PCA) to spatially illustrate the differentiation among sparrows using the ‘adegenet’ package (Jombart 2008; R Core Team 2019). An AMOVA was calculated in GenAlEx 6.501 using 9999 pairwise permutations of the data to evaluate differentiation among individual sparrows and subpopulations as well was the effective number of migrants (Nm) calculated globally. GenAlEx 6.501 was used to assess genetic differentiation among subpopulations A, B, C, D and E by comparing magnitudes of genetic differentiation with FST and comparing genetic differentiation without sampling bias or heterozygosity influence with DEST (Wright 1951; Jost 2008; Bird et al. 2011). Probabilities of pairwise comparisons were computed after 9999 permutations and alphas were adjusted for multiple comparisons using the sequential Holm-Bonferroni method (Holm 1979; Rice 1989).
To approximate the relatedness of sparrows within each subpopulation, mean Queller and Goodnight (1989) (Q-G) relatedness estimates were computed in GenAlEx with 9999 permutations and 9999 bootstrap repetitions to obtain 95% confidence intervals and determine statistical significance. GeneClass2.0 evaluated recent gene flow (up to two generations in the past), potential dispersal dynamics among subpopulations and approximate first-generation migrants (Piry et al. 2004). The Lh/Lmax test statistic compares the likelihood an individual’s genotype originated from the source subpopulation (Lh) to the maximum likelihood that the genotype can be found in any subpopulation (Lmax) (Paetkau et al. 2004). We used the prior probability of L = Lh/Lmax to determine migrant status with estimations using 10,000 MCMC trials and adjusted the alpha value according to pairwise comparisons (adjusted α = 0.01) (Paetkau et al. 2004). A conservative Bonferroni correction was applied if geneflow was high (Nm > 10) among subpopulations (adjusted α = 0.0009). BayesAss 1.3 software was also used to determine recent migration rates. The MCMC was run through a Bayesian estimator with one million burn-in steps for stabilization in 10 million iterations and sampling occurred every 1000 iterations (Wilson and Rannala 2003). The delta values for allele frequency, migration rate, and the level of inbreeding were tested from 0.10 to 0.65, in 0.5 increments, to mix the model for each iteration (Wilson and Rannala 2003).
Results
Mitochondrial and microsatellite genetic diversity
Feather samples were collected from June 2011 to June 2018 in management units A, B, C, D, and E. From DNA extractions of 178 sparrows, 114 DNA extractions were of high quality and yield. Extracted DNA using the modified PCI protocol yielded DNA quantities ranging from 3.1 to 301.8 ng/µL (mean = 20.4 ng/µL, standard error = 3.6 ng/µL). DNA from a total of 108 unique sparrows yielded successful and quality mitochondrial, microsatellite or both types of DNA for analyses (sample numbers and management units per marker given below). Due to the low quantity and quality DNA, variable numbers of sparrow samples were sequenced from the years 2011 (n = 1), 2014–2017 (n > 12 each year), and 2018 (n = 6) (Ntotal = 94). Samples successfully amplified for the mtDNA marker fragments of Cytb (936 basepairs; n = 37), D-loop (410 bp; n = 68), ND2 (984 bp; n = 36) and 16S (484 bp; n = 69) (Tables 1, S5, S6). We provide results for the Cytb and 16S markers online since sample sizes and genetic diversity were low (Tables S5, S6). Low differentiation among management units was identified, and therefore diversity parameters were analyzed with all units combined (Tables 1, S5). All ND2 sequences were of Haplotype 1 (GenBank reference MN263900), except for one sparrow from management unit B exhibited Haplotype 3 (GenBank reference MN263902); both ND2 haplotypes have been previously published for the species (Table S6) (Roeder et al. 2021). The D-loop locus also had one variable site and three haplotypes, dominated by haplotype 1 (86.9%, n = 60) across the management units and uncommon haplotypes in units C and E (Fig. 1b, Table S6). The D-loop dataset had higher nucleotide diversity with more nucleotide variation than ND2 (Tables 1, S6). A total of 11 sparrows had unique haplotypes in Cytb and/or D-loop loci (Fig. S2, Table S6). Haplotypes were submitted to GenBank (references OL790020- OL7900027, OL792049) and all sequences generated for all four markers are provided in Beaver et al. (2023).
The final microsatellite dataset included a total of 55 sparrows genotyped across 12 loci with < 7.5% allelic data missing overall and a genotype error rate of 0.9%. The 55 genotyped sparrows include 41 of the 94 sparrows sequenced for mtDNA and 14 additional sparrows that did not produce suitable mtDNA sequences. Genotyped sparrow samples were collected in 2011/2014–2015 (n = 1, each year), 2016 (n = 7), 2017 (n = 40), and 2018 (n = 5). Of the original 18 multiplexed loci, four loci were removed in the final dataset due to monomorphism (Asµ15) or inconsistent and poor-quality amplification leading to > 25% of samples missing data (Am12, Am14, ZoleF11). Additionally, two loci (ZoleG03 and Sosp13) were removed for evidence of null alleles (Table S3).
The remaining dataset of 12 loci was used in further analyses and did not show evidence of null alleles, stuttering errors or short allele dominance. The management unit’s sample sizes varied, with < 10 sparrows genotyped from management units A and D (Table 2). After correcting alpha using the Bonferroni method, all loci were found to be in Hardy–Weinberg equilibrium (adjusted α = 0.0042) and there was no evidence of linkage disequilibrium (adjusted α = 0.000758) among any pairwise comparisons of loci. Values for PIC ranged from 0.63 to 0.74 for individual subpopulations and averaged 0.74 with standard error ± 0.05 when all management units were treated as one, indicating loci can be informative even with low sample sizes for some units. All genotypes are accessible in Beaver et al. (2023).
Low levels of unique genetic diversity and inbreeding were observed utilizing microsatellite loci. No significant differences were found among management units in any measures of genetic diversity when randomized block ANOVA tests and Dunn’s tests alphas were adjusted for multiple comparisons (adjusted α > 0.005). Overall management units, the total number of alleles was 5.77 ± 0.23 (standard error, SE), while. similar levels of allelic richness were found in each management unit (Table 2). The number of private alleles was highest in management units B and E (AP = 6) and lowest for units A and D (AP = 1), with unit E in the middle (AP = 3). Expected heterozygosity was 0.71 (standard error = 0.02) averaged over all management units (Table 2). Low levels of inbreeding on average were observed across all management units (FIS = 0.02, Table 2). When compared to data of other subspecies, the Cape Sable subspecies had the lowest values for three genetic diversity parameters: number of alleles, allelic richness, and observed heterozygosity (Table 3). The Cape Sable subspecies had the second lowest expected heterozygosity values, second to the south Texas sparrows (A. m. cf. sennetti), and overall had similar genetic diversity values to the isolated Texas population (Table 3). Bottleneck software found no evidence of bottlenecks occurring when subpopulations were considered one population (Piry et al. 1999).
Generation length and age at first breeding was estimated from a total of 18 nestlings that were first captured and banded from June 2011–June 2019 and recaptured from May 2012–June 2020 (Table S4). The age at first breeding (F) for Cape Sable seaside sparrows was estimated between 310 and 710 days old (mean = 376.00, SE = 20.72). To calculate generation length (G), we used a subspecies-specific survival rate (S) reported by Boulton et al. (2009) at 0.59 (0.52–0.66), as well as an estimated longevity (L) reported by Klimkiewicz and Futcher (1989) for seaside sparrows in Florida at 8 years 11 months (8.92 years). Our mean estimate for F was 1.03 years which fell within the grouping of F = 0.5–1.5 provided in Bird et al. (2020) and corresponded to a 0.142 average z value. We calculated generation length for all three estimates of the survival rate resulting in an average generation length of GFLS = 2.35, minimum of GFL = 2.15, and maximum of GFLS = 2.67. The estimated effective population size for all samples combined based on the linkage disequilibrium method was 202.4 sparrows with a 95% confidence interval of 127.1–456.9 sparrows. Based on the estimated total population size in 2018 (total 3152 sparrows) and the estimated effective population size with a 95% confidence interval (202.4 sparrows, 127.1–456.9 sparrows), we found the ratio of NC:NE = 0.06, range 0.04–0.15 (Table 3) (U.S. Fish and Wildlife Service 2020).
Management unit connectivity
Bayesian, multivariate, and genetic distance analyses of microsatellite DNA reflected the results found with the mtDNA markers. Structure supported one inferred population as the most probable hypothesis with LnP(K) values greatest for K = 1, which ΔK cannot compute (Fig. S1). For reference, the K = 2 plot is provided in Fig. S1, although it was not selected as the most probable number of populations. The PCA explained 35.95% of the variation in the data set and showed no discernable differences among management units as indicated by the overlap of the 95% ellipses for each subpopulation (Fig. 2). An AMOVA found the highest variation within individuals (88%), then among individuals (11%), with a low global FST not statistically significant (FST = 0.008, P = 0.11) and a high effective number of migrants (Nm = 30.72). No significant differences were found in FST or DEST values when comparing subpopulations (Table S7; adjusted α = 0.005).

Microsatellite principal component analysis (PCA) presenting the first and second principal components to display 35.95% of the genetic variation in Cape Sable seaside sparrows, Florida, USA, 2011–2018. U.S. Fish and Wildlife defined subpopulations, or management units, are color-coded to match Fig. 1 and have different symbols: A (red square), B (blue dot), C (yellow triangle), D (green open rhombus) and E (grey asterisk). Individual sparrows are represented as symbols, with ellipses around 95% of the samples. A graphic summary of the eigenvalues is provided in the top right corner to explain that the most variation in the data is found in the first two principal components (black bars)
Fine-scale analyses utilizing relatedness values provided evidence for the overall connectivity among management units and potential skewed migration routes between different units. The within management unit average Q-G relatedness values were significantly higher than expected values for units A and B (Fig. 3; P = 0.003 and 0.012, respectively) (Queller and Goodnight 1989). However, since we have small sample sizes and the confidence limit includes zero, there is a possibility that significant relatedness is not present in management unit B. GeneClass2.0 identified one individual as having genotypes that significantly differed from their recorded management unit of origin (P = 0.008; adjusted α = 0.01). The one sparrow identified as a putative migrant was found in management unit B with the highest likelihood of migrating from unit E (Lhome/Lmax = 4.1). No individuals were identified as putative migrants when using the conservative Bonferroni correction (adjusted α = 0.0009). Another sparrow identified as a putative migrant was first captured as a nestling in 2012 at management unit B and was recaptured in unit D two years later, but feather samples were only collected during recapture and genetic matching was not possible. Efforts were made to estimate recent migration rates among subpopulations using BayesAss 1.3, but the delta parameter for the F-statistic was not able to be calculated within the range of acceptance (20–60%) or within the recommended range for delta parameters (< 0.45); likely an artefact of the limited samples sizes (Wilson and Rannala 2003; Woltmann et al. 2014).

A summary of the U.S. Fish and Wildlife defined subpopulations, or management units, intra-unit (A–E) genetic mean relatedness values for Cape Sable seaside sparrow units (open circles) from Florida, USA, 2011–2018. Error bars represent standard deviations. The dashed lines indicate the upper and lower bounds of the 95% confidence interval for each management unit. Mean relatedness for unit A was statistically significant (P = 0.003) and the upper and lower confidence limits did not include zero as indicated by an asterisk (*) above the error bar
Discussion
This is the first study to assess the unique diversity across the Cape Sable seaside sparrow management units and provide a comprehensive genetic profile indicating a panmictic population, although sample sizes were limited. This is also the first seaside sparrow genetic diversity study to date, to assess other mtDNA markers (i.e., Cytb, D-loop, 16S) along with ND2, which indicated low diversity of matriarchal ancestry in the Cape Sable seaside sparrow subspecies. The nuclear microsatellite analyses (e.g., Bayesian, multivariate, distance statistics) consistently identified all management units as one metapopulation with overall low genetic diversity. The management units east of the Shark River Slough (B, C, D and E) are likely to be more interconnected with each other than the A unit to the west based on relatedness and migration estimates by genetic analyses and mark-recapture observations. Cape Sable seaside sparrow management units (previously referred to as ‘subpopulations’) are managed separately and therefore, some baseline genetic diversity metrics were provided at the management unit level for future comparisons (e.g., private alleles or unique genetic diversity to each management unit and relatedness).
Comparison of genetic diversity to other subspecies
The two ND2 haplotypes in the Cape Sable seaside sparrow management units A and B matched Haplotype 1 found in almost every subspecies previously sampled, and Haplotype 3 from unit B was found in populations of Scott’s seaside sparrows (A. m. peninsulae) along the Gulf of Mexico coast and MacGillivray’s (A. m. macgillivraii)/Northern (A. m. maritima) seaside sparrows along the Atlantic coast (Woltmann et al. 2014; Roeder et al. 2021). Among 36 Cape Sable seaside sparrows, we observed a lower ND2 haplotype frequency than that found in other Florida subspecies, such as two haplotypes among 11 Wakulla seaside sparrows (A. m. juncicola), and three haplotypes among 12 Scott’s seaside sparrows (Woltmann et al. 2014). The low numbers of mtDNA haplotypes may indicate low historic diversity from post-glaciation dispersal of the species or loss of haplotypes during isolation and population size reduction events (Zink and Dittmann 1993; Roeder et al. 2021). For completeness, more and better-quality Cape Sable seaside sparrow DNA samples are needed to sequence the ND2 locus from the C, D, E and F subpopulations to examine the genetic diversity more comprehensively. Of the sequenced mtDNA loci, only the ND2 locus is published in other subspecies to allow for comparison.
Cape Sable seaside sparrows had the lowest relative average nuclear genetic diversity compared to other subspecies (although note the use of different microsatellite panels and lack of locus-specific information for other subspecies; Davis et al. 2021; Roeder et al. 2021). The results in the present study are supported by a species-wide RAD-Seq study finding the lowest subspecies diversity in two Cape Sable seaside sparrow samples compared to other subspecies (Davis et al. 2021). Both Davis et al. (2021) and this study have low samples sizes (< 10 sparrows in some groupings) leading to potential bias in datasets, however, due to poor quality DNA yield, these studies provide the only genetic profiles published to date. The Cape Sable seaside sparrows have the lowest genetic diversity values in most metrics reported within the species’ complex and may be important to consider in management activities, especially in light of the rapid extinction of the Dusky Seaside sparrow (A. m. nigrescens) (Avise and Nelson 1989; Zink and Kale 1995; Woltmann et al. 2014; Davis et al. 2021).
Comparison of genetic data to annual mark-recapture-survey observations
Observed trends in demographic surveys can provide context for genetic data on migration and relatedness across management units. The observed genetic connectivity from subpopulation E to B is supported by leg banding mark-recapture events documented between 2007 and 2017 with dispersal from subpopulation B to C, B to D, C to B, D to B and F to B, and frequent dispersal activity from subpopulation E to C (Virzi et al. 2018; Virzi and Tafoya 2020, unpublished data from T. Virzi). Frequent and recent immigration can increase the subpopulation size and potentially reduce genetic relatedness values, likely reflected in the values we found in management unit D as a result of the influx of immigrants observed in recent years (Virzi and Tafoya 2020). Conversely, high self-recruitment, natal site fidelity and a lack of immigration can all increase relatedness values. This was observed in the geographically distant management unit A, where mark-recapture data documented high self-recruitment between 2008 and 2020, and no dispersal events (Virzi and Tafoya 2020). Even though the maximum known distance of sparrow dispersal is > 30 km, which is enough to connect all subpopulations in a stepwise pattern, these long-distance dispersal events have not been observed in the current survey efforts over the past 10 years (Van Houtan et al. 2010; Virzi and Tafoya 2020). Since the most important protection of Cape Sable seaside sparrows from catastrophic events was identified as maintaining breeding populations across a wide geographic area (U.S. Fish and Wildlife Service 2020), the potential loss of management unit A due to low immigration and geographic separation is important to consider.
Observations at annual surveys suggest that management unit A has undergone multiple population bottlenecks, which can quickly eliminate unique genetic diversity and decrease the number of breeding sparrows in a population, further reducing genetic variation. Although no bottlenecks were detected by genetic analyses, demographic bottlenecks were suspected. Total population size declines were detected during annual surveys beginning in the early 1990s after water pattern changes (Curnutt et al. 1998) and also in 2018 when the total population size in management unit A declined by 75% after Hurricane Irma, leaving a single female likely responsible for successful breeding each subsequent season (Virzi and Tafoya 2020). Furthermore, recent wildfires in management unit A (e.g., the Guava Fire in 2020) likely resulted in unsuccessful breeding seasons of sparrows as the landscape recovered (La Puma et al. 2007; Virzi and Tafoya 2021). It is important to note that the observed demographic bottlenecks refer to the total population size and do not detail how the breeding population was impacted (relationship between breeding, NE, and total population size, NC, discussed below).
Genetic diversity and conservation implications for Cape Sable
Deficient levels of contemporary geneflow between management units can increase the likelihood of inbreeding, and lead to the need for genetic rescue through management interventions (Zink and Kale 1995; Pimm et al. 2006). Previous studies of fragmented and small populations suggest that implementing genetic rescue based on available genetic data can lead to improved conservation outcomes (Hufbauer et al. 2015; Ralls et al. 2020). Population units with high levels of inbreeding (FIS > 0.1) may warrant management intervention (e.g., translocations or augmentation) (Ralls et al. 2020). Inbreeding coefficients reported here represent a snapshot at the time of sample collection (prior to 2017–2018) but may need to be re-evaluated given the change in environmental conditions in more recent years (further discussed in the ‘Future efforts’ section).
The ability to estimate population vulnerability and resiliency to stochastic events is crucial when assessing the application of potential management interventions. The generation length has never been published explicitly for Cape Sable seaside sparrows but could serve as a proxy to estimate population resiliency. The generation length reported here (2.15–2.67 years) is similar to most bird species assessed in Bird et al. (2020). Based on the life history parameters used to calculate generation length (e.g., age of first reproduction, maximum longevity and annual adult survival), population trends for the Cape Sable seaside sparrows can be adequately assessed within the 10 year period for precautionary threat assessment used by the International Union for Conservation of Nature (IUCN) Red List criterion A (IUCN 2012).
The ratio of breeding population size (or NE) to total population size (or NC) can be used as an indicator of genetic loss since neutral loci can lose genetic variation at a rate of 1/(2 × NE) per generation on average (Hoban et al. 2020). The overall ratio observed for Cape Sable seaside sparrows was less than the average for stable wild populations (NE:NC = 0.1) suggesting limited genetic diversity, however, the 95% confidence interval does include the stable range (NE:NC = 0.04–0.14) (Frankham 1995; Palstra and Ruzzante 2008; Hoban et al. 2020, 2021; Laikre et al. 2021). Hoban et al. (2021) state, “Ne > 500 is an appropriate, well-accepted threshold for maintaining genetic diversity in diverse organisms” and “Ne > 5000 is a useful threshold for guiding genetic diversity conservation”. As observed through annual mark-recapture events, Cape Sable seaside sparrows have experienced fluctuating population sizes (argued the most impactful factor, Frankham 1995; Vucetich et al. 1997), lower female survival rates, strongly male-skewed sex ratios and overlapping generations, all of which can decrease the breeding population size (Nott et al. 1998; Jenkins et al. 2003; Boulton et al. 2009; Virzi et al. 2018). Conversely, an increase in population census size and subsequent breeding population sizes, can lead to additional genetic mutation or immigration events which could increase genetic diversity among management units. By supporting more consistent breeding and population growth, a larger census population size could help to prevent genetic diversity loss during stochastic events (Nott et al. 1998; Pimm and Bass 2002; Jenkins et al. 2003; Ross et al. 2003; Basier et al. 2008; Boulton et al. 2009). By assessing the breeding population size and inbreeding levels, the population health can be monitored to avoid the need for taking dire genetic rescue efforts, as occurred with the extinction of the Dusky seaside sparrow (A. m. nigrescens), the closest geographic population to the Cape Sable seaside sparrow (Avise and Nelson 1989; Hill 1993; Zink and Kale 1995; Baalsrud et al. 2014).
Future efforts to advance Cape Sable seaside sparrow research
Past habitat alterations and water management have disrupted Cape Sable seaside sparrow management units and likely influenced genetic diversity. Since all six management units are in low-lying areas of southern Florida, rising sea levels and saltwater intrusion are the highest predicted challenges for the future (Virzi et al. 2018). Management unit B is the farthest south, closest to the ocean, and is projected to be the first vulnerable to saltwater intrusion by the U.S. Army Corps of Engineers (U.S. Fish and Wildlife Service 2019; Benscoter et al. 2021; Romañach et al. 2023). Management unit B also has the largest surface area and the highest level of unique genetic diversity (equal to unit E), potentially containing a preponderance of the current genetic diversity present for this subspecies. The management unit relationships we describe could inform efforts to increase population resiliency prior to the potential effects of sea level rise and other climate change impacts (Roberts et al. 2019; Romañach et al. 2023).
Future genetic monitoring of the subspecies could benefit from exploring other sampling techniques (e.g., Cambrone et al. 2022) since limited DNA was extracted from a restricted number of small feathers. A limitation of this study is that nuclear microsatellites are neutral loci and therefore, natural selection or local adaptation cannot be identified using these markers (Dauphin et al. 2020; Han et al. 2020). Future efforts could employ high density genome-wide nuclear assessments [e.g., single nucleotide polymorphisms (SNP) methods] to assess the contemporary genetic vulnerability and adaptation of sparrows in each management unit and to investigate temporal changes in genetic diversity prior to, and after, population declines or translocations (Miller et al. 2012; Dresser et al. 2017; Card et al. 2018). However, the DNA quality and quantity from sparrow feathers extracted here using here optimized and modified methods, precluded RADseq analyses of SNPs in a separate study except for two samples (Davis et al. 2021). Obtaining quality blood samples, as used in previous seaside sparrow genetic studies, is difficult for this endangered sparrow but could assist with SNP analyses or improve the quality and quantity of mitochondrial and microsatellite data (Woltmann et al. 2014; Carroll et al. 2018; Davis et al. 2021; Enloe et al. 2021). The first neutral nuclear genetic diversity profile for the subspecies provided here can serve as a baseline comparison for future studies. Maintaining or increasing diversity in the endangered Cape Sable seaside sparrow population can help to encourage resiliency in the changing Greater Everglades ecosystem environment.
Data availability
Haplotype data are publicly accessible on NCBI GenBank (references OL790020, OL7900027, OL792049). All mitochondrial DNA sequences and microsatellite allelic data are provided in full at https://doi.org/10.5066/P9NYGMI1.
References
Alström P, Olsson U, Lei F, Wang HT, Gao W, Sundberg P (2008) Phylogeny and classification of the Old World Emberizini (Aves, Passeriformes). Mol Phylogen Evol 47(3):960–973
Avise JC, Nelson WS (1989) Molecular genetic relationships of the extinct dusky seaside sparrow. Science 243:646–648
Baalsrud HT, Saether BE, Hagen IJ, Myhre AM, Ringsby TH, Parn H, Jensen H (2014) Effects of population characteristics and structure on estimates of effective population size in a house sparrow metapopulation. Mol Ecol 23(11):2653–2668. https://doi.org/10.1111/mec.12770
Basier RL, Boulton RL, Lockwood JL (2008) Influence of water depth on nest success of the endangered Cape Sable seaside sparrow in the Florida Everglades. Anim Conserv 11:190–197
Bass OL, Kushlan JA (1982) Status of the Cape Sable sparrow. Report T-672. National Park Service, South Florida Research Center, Everglades National Park; Homestead, Florida
Beaver CE, Virzi T, Hunter ME (2023) Mitochondrial and microsatellite genetic data from Cape Sable seaside sparrow feather samples collected 2011–2018: U.S. Geological Survey data release.
Benscoter AM, Haider SM, Guilbeau KG, Romañach SS (2021) Knowledge synthesis of Cape Sable seaside sparrow science. Report to the U.S. Fish and Wildlife Service
Benscoter AM, Romañach SS (2022) Endangered Cape Sable seaside sparrow ecology: actions towards recovery through landscape-scale ecosystem restoration. Endangered Species Research 49:199–215. https://doi.org/10.3354/esr01212
Bibby CJ, Burgess ND, Hill DA (1992) Bird census techniques. Academic Press, New York
Bird CE, Karl SA, Smouse PE, Toonen RJ (2011) Detecting and measuring genetic differentiation. In: Held C, Koenemann S, Schubart CD (eds) Phylogeography and population genetics in Crustacea, vol 19. CRC Press, Boca Raton
Bird JP, Martin R, Akcakaya HR, Gilroy J, Burfield IJ, Garnett ST, Symes A, Taylor J, Sekercioglu CH, Butchart SHM (2020) Generation lengths of the world’s birds and their implications for extinction risk. Conserv Biol 34(5):1252–1261. https://doi.org/10.1111/cobi.13486
Boulton RL, Lockwood JL, Davis MJ, Pedziwilk A, Boadway KA, Boadway JJT, Okines D, Pimm SL (2009) Endangered Cape Sable seaside sparrow survival. J Wildl Manage 73(4):530–537. https://doi.org/10.2193/2007-467
Bulgin NL, Gibbs HL, Vickery P, Baker AJ (2003) Ancestral polymorphisms in genetic markers obscure detection of evolutionarily distinct populations in the endangered Florida grasshopper sparrow (Ammodramus savannarum floridanus). Mol Ecol 12(4):831–844. https://doi.org/10.1046/j.1365-294X.2003.01774.x
Cambrone C, Motreuil S, Reyes FO, Landestoy MA, Cézilly F, Bezault E (2022) Obtaining DNA samples from sensitive and endangered bird species: a comparison of saliva and blood samples. Ardeola. https://doi.org/10.13157/arla.69.2.2022.sc1
Card DC, Perry BW, Adams RH, Schield DR, Young AS, Andrew AL, Jezkova T, Pasquesi GIM, Hales NR, Walsh MR, Rochford MR, Mazzotti FJ, Hart KM, Hunter ME, Castoe TA (2018) Novel ecological and climatic conditions drive rapid adaptation in invasive Florida Burmese pythons. Mol Ecol 27(23):4744–4757. https://doi.org/10.1111/mec.14885
Carroll EL, Bruford MW, DeWoody JA, Leroy G, Strand A, Waits L, Wang J (2018) Genetic and genomic monitoring with minimally invasive sampling methods. Evol Appl 11(7):1094–1119. https://doi.org/10.1111/eva.12600
Curnutt JL, Mayer AL, Brooks TM, Manne L, Bass OL, Fleming DM, Nott MP, Pimm SL (1998) Population dynamics of the endangered Cape Sable seaside-sparrow. Anim Conserv 1(1):11–21. https://doi.org/10.1111/j.1469-1795.1998.tb00221.x
Dauphin B, Wuest RO, Brodbeck S, Zoller S, Fischer MC, Holderegger R, Gugerli F, Rellstab C (2020) Disentangling the effects of geographic peripherality and habitat suitability on neutral and adaptive genetic variation in Swiss stone pine. Mol Ecol 29(11):1972–1989. https://doi.org/10.1111/mec.15467
Davis KEC, Settlecowski AE, Roeder MR, Enloe C, Virzi T, Hunter ME, Woltmann S, Taylor SS (2021) Range-wide population genetic analysis of seaside sparrows (Ammospiza maritima) supports at least five distinct population segments that do not align with current subspecies descriptions. Ornithol Appl. https://doi.org/10.1093/ornithapp/duab019
Dean TF, Morrison JL (2001) Non-breeding season ecology of the Cape Sable seaside sparrow (Ammodramus maritimus mirabilis).
Dinno A (2017) Dunn’s test of multiple comparisons using rank sums.
Dresser CM, Ogle RM, Fitzpatrick BM (2017) Genome scale assessment of a species translocation program. Conserv Genet 18(5):1191–1199. https://doi.org/10.1007/s10592-017-0970-6
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Enloe C, Cox WA, Pandey A, Taylor SS, Woltmann S, Kimball RT (2021) Genome-wide assessment of population structure in Florida’s coastal seaside sparrows. Conserv Genet. https://doi.org/10.1007/s10592-021-01411-9
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Flanagan SP, Forester BR, Latch EK, Aitken SN, Hoban S (2018) Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evol Appl 11(7):1035–1052. https://doi.org/10.1111/eva.12569
Foran DR, Fischer AB, Stoloff ME (2015) A comparison of mitochondrial DNA amplification strategies for species identification. J Forensic Investig. https://doi.org/10.13188/2330-0396.1000025
Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66(2):95–107
Gilroy JJ, Virzi T, Boulton RL, Lockwood JL (2012) A new approach to the “apparent survival” problem: estimating true survival rates from mark–recapture studies. Ecology 93(7):1509–1516
Goudet J (2002) FSTAT version 2.9.3.2. A program to estimate and test gene diversities and fixation indices. Institute of Ecology, Lausanne
Griscom L (1944) A second revision of the seaside sparrows occasional papers of the museum of natural science. La State Univ 1(19):1
Han F, Jamsandekar M, Pettersson ME, Su L, Fuentes-Pardo AP, Davis BW, Bekkevold D, Berg F, Casini M, Dahle G, Farrell ED, Folkvord A, Andersson L (2020) Ecological adaptation in Atlantic herring is associated with large shifts in allele frequencies at hundreds of loci. Elife. https://doi.org/10.7554/eLife.61076
Hill KD (1993) The Endangered species act: what do we mean by species. HeinOnline 20(2):239–264
Hill CE, Tomko S, Hagen C, Schable NA, Glenn TC (2008) Novel microsatellite markers for the saltmarsh sharp-tailed sparrow, Ammodramus caudacutus (Aves: Passeriformes). Mol Ecol Resour 8(1):113–115. https://doi.org/10.1111/j.1471-8286.2007.01885.x
Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, Sjögren-Gulve P, Segelbacher G, Vernesi C, Aitken S, Bertola LD, Bloomer P, Breed M, Rodríguez-Correa H, Funk WC, Grueber CE, Hunter ME, Jaffe R, Liggins L, Mergeay J, Moharrek F, O’Brien D, Ogden R, Palma-Silva C, Pierson J, Ramakrishnan U, Simo-Droissart M, Tani N, Waits L, Laikre L (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biol Conserv. https://doi.org/10.1016/j.biocon.2020.108654
Hoban S, Paz-Vinas I, Aitken S, Bertola LD, Breed MF, Bruford MW, Funk WC, Grueber CE, Heuertz M, Hohenlohe P, Hunter ME, Jaffé R, Fernandes ML, Mergeay J, Moharrek F, O’Brien D, Segelbacher G, Vernesi C, Waits L, Laikre L (2021) Effective population size remains a suitable, pragmatic indicator of genetic diversity for all species, including forest trees. Biol Conserv. https://doi.org/10.1016/j.biocon.2020.108906
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6(2):65–70
Hufbauer RA, Szűcs M, Kasyon E, Youngberg C, Koontz MJ, Richards CT, Melbourne BA (2015) Three types of rescue can avert extinction in a changing environment. Proc Natl Acad Sci 112(33):10557–10562
IUCN (International Union for Conservation of Nature) (2012) IUCN red list categories & criteria. Version 3.1. IUCN, Gland, Switzerland
Jenkins CN, Powell RD, Bass OL, Pimm SL (2003) Demonstrating the destruction of the habitat of the Cape Sable seaside sparrow (Ammodramus maritimus mirabilis). Anim Conserv 6(1):29–38. https://doi.org/10.1017/s1367943003003056
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res. https://doi.org/10.1093/nar/gkn201
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kale HWI (1983) Distribution, habitat and status of breeding seaside sparrows in Florida. In: Quay TLFJ, Potter EF, Robbins CS (eds) The seaside sparrow, its biology and management. Occasional Papers of the North Carolina Biological Survey, Raleigh, pp 41–48
Kalinowski S, Taper M, Marshall T (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol Notes 16:1006–1099
Klimkiewicz MK, Futcher AG (1989) Longevity records of North American birds: supplement I. J Field Ornithol 60(4):469–494
La Puma DA, Lockwood JL, Davis MJ (2007) Endangered species management requires a new look at the benefit of fire: the Cape Sable seaside sparrow in the Everglades ecosystem. Biol Conserv 136(3):398–407. https://doi.org/10.1016/j.biocon.2006.12.005
Laikre L, Hohenlohe PA, Allendorf FW, Bertola LD, Breed MF, Bruford MW, Funk WC, Gajardo G, González-Rodríguez A, Grueber CE, Hedrick PW, Heuertz M, Hunter ME, Johannesson K, Liggins L, MacDonald AJ, Mergeay J, Moharrek F, O’Brien D, Ogden R, Orozco-terWengel P, Palma-Silva C, Pierson J, Paz-Vinas I, Russo I-RM, Ryman N, Segelbacher G, Sjögren-Gulve P, Waits LP, Vernesi C, Hoban S (2021) Authors’ reply to letter to the editor: continued improvement to genetic diversity indicator for CBD. Conserv Genet. https://doi.org/10.1007/s10592-021-01359-w
Lehmicke AJJ, Berry BE, Shamblin BM, Lennon DM, Woodrey MS, Cooper RJ, Nairn CJ (2012) Isolation and characterization of tetranucleotide microsatellite loci from the seaside sparrow (Ammodramus maritimus). Conserv Genet Resour 4(4):881–884. https://doi.org/10.1007/s12686-012-9664-z
Leigh JW, Bryant D, Nakagawa S (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110–1116. https://doi.org/10.1111/2041-210x.12410
Leigh DM, van Rees CB, Millette KL, Breed MF, Schmidt C, Bertola LD, Hand BK, Hunter ME, Jensen EL, Kershaw F, Liggins L, Luikart G, Manel S, Mergeay J, Miller JM, Segelbacher G, Hoban S, Paz-Vinas I (2021) Opportunities and challenges of macrogenetic studies. Nat Rev Genet 22(12):791–807. https://doi.org/10.1038/s41576-021-00394-0
Lockwood JL, Fenn KH, Caudill JM, Okines D, Bass OL, Duncan JR, Pimm SL (2001) The implications of Cape Sable seaside sparrow demography for Everglades restoration. Anim Conserv 4(3):275–281. https://doi.org/10.1017/s1367943001001329
Lockwood JL, Ross MS, Sah JP (2003) Smoke on the water: the interplay of fire and water flow on Everglades restoration. Front Ecol Environ 9(1):462–468
Maddison WP, Maddison DR (2018) Mesquite: a modular system for evolutionary analysis.
McDonald MV (1988) Status survey of two Florida seaside sparrows and taxonomic review of the seaside sparrow assemblage. Jacksonville, Florida: Florida Cooperative Fish and Wildlife Research Unit, School of Forestry and Conservation, University of Florida Technical Report No. 32
Mila B, McCormack JE, Castaneda G, Wayne RK, Smith TB (2007) Recent postglacial range expansion drives the rapid diversification of a songbird lineage in the genus Junco. Proc Royal Soc Biol Sci 274(1626):2653–2660. https://doi.org/10.1098/rspb.2007.0852
Miller JM, Poissant J, Hogg JT, Coltman DW (2012) Genomic consequences of genetic rescue in an insular population of bighorn sheep (Ovis canadensis). Mol Ecol 21(7):1583–1596. https://doi.org/10.1111/j.1365-294X.2011.05427.x
Nelson WS, Dean T, Avise JC (2000) Matrilineal history of the endangered Cape Sable seaside sparrow inferred from mitochondrial DNA polymorphism. Mol Ecol 9(6):809–813
Noecker RJ, Environment and Natural Resources Policy Division (1998) Endangered species list revisions: a summary of delisting and downlisting. Congressional Research Service, Library of Congress
Nott MP, Bass OL, Fleming DM, Killeffer SE, Fraley N, Manne L, Curnutt JL, Brooks TM, Powell R, Pimm SL (1998) Water levels, rapid vegetational changes and the endangered Cape Sable seaside-sparrow. Anim Conserv 1(1):23–32. https://doi.org/10.1111/j.1469-1795.1998.tb00222.x
Ottewell K, Byrne M (2022) Conservation genetics for management of threatened plant and animal species. Diversity. https://doi.org/10.3390/d14040251
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13(1):55–65. https://doi.org/10.1046/j.1365-294X.2003.02008.x
Palstra FP, Ruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17(15):3428–3447. https://doi.org/10.1111/j.1365-294x.2008.03842.x
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peel D, Ovenden JR, Peel SL (2004) NeEstimator: software for estimating effective population size, Version 1.3. Queensland Government, Department of Primary Industries and Fisheries
Pimm SL, Bass OL Jr (2002) Range-wide risks to large populations: the Cape Sable seaside sparrow as a case history. In: Beissinger SR, McCullough DL (eds) Population viability analysis. The University of Chicago Press, Chicago, pp 406–424
Pimm SL, Lockwood JL, Jenkins CN, Curnutt JL, Nott MP, Powell RD, Bass OL Jr (2002) Sparrow in the grass: a report on the first 10 years of research on the Cape Sable seaside sparrow. Everglades National Park, Homestead
Pimm SL, Dollar L, Bass OL (2006) The genetic rescue of the Florida panther. Anim Conserv 9(2):115–122. https://doi.org/10.1111/j.1469-1795.2005.00010.x
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90(4):502–503
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) Geneclass2: a software for genetic assignment and first-generation migrant detection. J Hered 95(6):536–539. https://doi.org/10.1093/jhered/esh074
Platt WJ, Beckage B, Doren RF, Slater HH (2002) Interactions of large-scale disturbances: prior fire regimes and hurricane mortality of savanna pines. Ecology 83(6):1566–1572
Poesel A, Gibbs HL, Nelson DA (2009) Twenty-one novel microsatellite DNA loci isolated from the Puget Sound white-crowned sparrow, Zonotrichia Leucophrys Pugetensis. Mol Ecol Resour 9(3):795–798. https://doi.org/10.1111/j.1755-0998.2008.02182.x
Queller DC, Goodnight GK (1989) Estimating relatedness using genetic markers. Evolution 43(2):258–275
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Radford JQ, Amos N, Harrisson K, Sunnucks P, Pavlova A (2021) Functional connectivity and population persistence in woodland birds: insights for management from a multi-species conservation genetics study. Emu-Austral Ornithol 121:1–13
Ralls K, Sunnucks P, Lacy RC, Frankham R (2020) Genetic rescue: a critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biol Conserv 251:108784
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Remsen JJV (2010) Subspecies as a meaningful taxonomic rank in avian classification. Ornithol Monogr 67(1):62–78. https://doi.org/10.1525/om.2010.67.1.62
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43(1):223–225
Robbins CS (1983) Distribution and migration of Seaside Sparrows. In: Quay TLFJ, Potter EF, Robbins CS (eds) The Seaside Sparrow, its biology and management. Occasional Papers of the North Carolina Biological Survey, Raleigh, pp 31–39
Roberts SG, Longenecker RA, Etterson MA, Elphick CS, Olsen BJ, Shriver WG (2019) Preventing local extinctions of tidal marsh endemic seaside sparrows and saltmarsh sparrows in eastern North America. Condor. https://doi.org/10.1093/condor/duy024
Roeder MR, Hill CE, Elphick CS, Conway M, Kocek AR, Tegeler A, Woltmann S (2021) Genetic data disagree with described subspecies ranges for seaside sparrows on the Atlantic coast. Ornithol Appl. https://doi.org/10.1093/ornithapp/duab011
Roman CT, Aumen NG, Trexler JC, Fennema RJ, Loftus WF, Soukup MA (1994) Hurricane Andrew’s impact on freshwater resources: water quality, so important to defining the Everglades’ unique ecological composition, appears little affected. Bioscience 44(4):247–255
Romañach SS, Haider SM, Benscoter AM (2023) Sea level rise may pose conservation challenges for the endangered Cape Sable seaside sparrow. Front Ecol Evol. https://doi.org/10.3389/fevo.2022.1085970
Ross MS, Sah JP, Ruiz PL, Jones DT, Cooley H, Travieso R, Snyder JR, Schaeffer C (2003) Effect of hydrologic restoration on the habitat of the Cape Sable seaside sparrow, annual report of 2002–2003. SERC Research Reports
Ross JD, Bouzat JL (2014) Genetic and morphometric diversity in the Lark Sparrow (Chondestes grammacus) suggest discontinuous clinal variation across major breeding regions associated with previously characterized subspecies. Auk 131(3):298–313. https://doi.org/10.1642/auk-13-246.1
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19(18):2496–2497. https://doi.org/10.1093/bioinformatics/btg359
Ryan RM, Woltmann S (2023) Variation in song between coastal lineages of seaside sparrow (Ammospiza maritima). Wilson J Ornithol. https://doi.org/10.1676/21-00073
Sah JP, Ross MS, Ruiz PL, Snyder JR, Rodriguez D, Hilton WT (2011) Cape Sable seaside sparrow habitat monitoring and assessment—2010 final report. SERC Research Reports
Sah JP, Snyder JR, Ross MS, Stoffella S, Constant B, Castaneda S, Pulido C (2021) Evaluation of vegetation response to changes in hydrologic parameters within Cape Sable seaside sparrow habitat, Everglades National Park, Florida Annual report—2020 (Year 4). Report submitted to U.S. Army–ERDC, Vicksburg, MS and South Florida Natural Resources Center, Everglades & Dry Tortugas National Parks, Homestead
Sambrook J, Fritsch E, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sardell RJ, Keller LF, Arcese P, Bucher T, Reid JM (2010) Comprehensive paternity assignment: genotype, spatial location and social status in song sparrows, Melospiza Melodia. Mol Ecol 19(19):4352–4364. https://doi.org/10.1111/j.1365-294X.2010.04805.x
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Linn Soc 68:41–55
Thompson LM, Thurman LL, Cook CN, Beever EA, Sgrò CM, Battles A, Botero CA, Gross JE, Hall KR, Hendry AP, Hoffmann AA, Hoving C, LeDee OE, Mengelt C, Nicotra AB, Niver RA, Pérez-Jvostov F, Quiñones RM, Schuurman GW, Schwartz MK, Szymanski J, Whiteley A (2023) Connecting research and practice to enhance the evolutionary potential of species under climate change. Conserv Sci Pract. https://doi.org/10.1111/csp2.12855
U.S. Fish and Wildlife Service (2006) Endangered and threatened wildlife and plants; critical habitat designation for the Cape Sable seaside sparrow proposed rule. Fed Reg 71:63980–64002. https://www.govinfo.gov/content/pkg/FR-2006-10-31/pdf/06-8930.pdf
U.S. Fish and Wildlife Service (2019) Recovery plan amendment for Cape Sable seaside sparrow (Ammodramus maritimus mirabilis) [Draft version]. U.S Fish and Wildlife Service, Atlanta
U.S. Fish and Wildlife Service (2020) Biological opinion for the combined operational plan. U.S Fish and Wildlife Service, Vero Beach
Van Houtan KS, Bass OL Jr, Lockwood J, Pimm SL (2010) Importance of estimating dispersal for endangered bird management. Conserv Lett 3(4):260–266. https://doi.org/10.1111/j.1755-263X.2010.00108.x
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Virzi T, Davis MJ, Slater G (2018) Recovery of Cape Sable seaside sparrow—subpopulation A. Annual Report to the U.S Fish and Wildlife Service, Vero Beach
Virzi T, Tafoya D (2020) Recovery of Cape Sable seaside sparrow. Report to the United States fish and wildlife service (South Florida Ecological Services Field Office), Vero Beach, Florida, USA and National Park Service (Everglades National Park), Homestead, Florida
Virzi T, Tafoya D (2021) Recovery of Cape Sable seaside sparrow. Annual report. Submitted to U.S. Fish and Wildlife Service, South Florida Ecological Services Office, and National Park Service, Everglades National Park. Conservation InSight, Happy Valley
Vucetich JA, Waite TA, Nunney L (1997) Fluctuating population size and the ratio of effective to census population size. Evolution 51(6):2017–2021
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163(3):1177–1191
Winker K (2010) Subspecies represent geographically partitioned variation, a gold mine of evolutionary biology and a challenge for conservation. Ornithol Monogr 67(1):6–23. https://doi.org/10.1525/om.2010.67.1.6
Woltmann S, Stouffer PC, Bergeon Burns CM, Woodrey MS, Cashner MF, Taylor SS (2014) Population genetics of seaside sparrow (Ammodramus maritimus) subspecies along the gulf of Mexico. PLoS ONE 9(11):e112739. https://doi.org/10.1371/journal.pone.0112739
Woltmann S, Roeder M, Xiong A, Hill C, Conway MH (2019) Additional notes on Texas seaside sparrows (Ammospiza maritima sennetti): an update to Griscom 1948. Wilson J Ornithol 131(4):750–757
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Zink RM, Dittmann DL (1993) Gene flow, refugia, and evolution of geographic variation in the song sparrow (Melospiza melodia). Evolution 47(3):717–729
Zink RM, Kale HW (1995) Conservation genetics of the extinct dusky seaside sparrow Ammodramus maritimus nigrescens. Biol Conserv 74:69–71
Acknowledgements
We are grateful to M. Davis for assisting with field work and collecting samples. We are also grateful to Dr. M. Jubair, and B. Caton and M. Giles for assisting with lab work and analyses. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U. S. Government.
Funding
This project was funded in significant part by the U. S. Geological Survey’s Greater Everglades Priority Ecosystems Science (GEPES) Program.
Author information
Authors and Affiliations
Contributions
MH and TV developed the project and acquired funding. TV completed field work and collected samples. CB and MH collected and processed data for the project in the laboratory. MH advised while CB calculated and performed data analyses. All authors assisted with data interpretation, biological, and ecological relevance. CB and MH prepared the manuscript for submission. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Feather samples were collected from juvenile and adult sparrows authorized under Federal Bird Banding Permit #23166 and Native Endangered Species Recovery Permit #TE075916 issued by the U.S. Department of the Interior. These permits ensure the study plan was approved by the U.S. Department of the Interior and animals were treated humanely and ethically.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beaver, C.E., Virzi, T. & Hunter, M.E. Genetic analysis of federally endangered Cape Sable seaside sparrow subpopulations in the Greater Everglades, USA. Conserv Genet 25, 101–116 (2024). https://doi.org/10.1007/s10592-023-01551-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01551-0