
Abstract
Australian arboreal mammals are experiencing significant population declines, particularly due to land clearing and resulting habitat fragmentation. The squirrel glider, Petaurus norfolcensis, is a threatened species in New South Wales, with a stronghold population in the Lake Macquarie Local Government Area (LGA) where fragmentation due to urbanization is an ongoing problem for the species conservation. Here we report on the use of squirrel glider mitochondrial (385 bp cytochrome b gene, 70 individuals) and nuclear DNA (6,834 SNPs, 87 individuals) markers to assess their population genetic structure and connectivity across 14 locations sampled in the Lake Macquarie LGA. The mitochondrial DNA sequences detected evidence of a historical genetic bottleneck, while the genome-wide SNPs detected significant population structure in the Lake Macquarie squirrel glider populations at scales as fine as one kilometer. There was no evidence of inbreeding within patches, however there were clear effects of habitat fragmentation and biogeographical barriers on gene flow. A least cost path analysis identified thin linear corridors that have high priority for conservation. These areas should be protected to avoid further isolation of squirrel glider populations and the loss of genetic diversity through genetic drift.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The hollow-dependent, arboreal squirrel glider (Petaurus norfolcensis, Kerr, 1792) is an Australian marsupial that forms monogamous social groups (Sharpe and Goldingay 2007), and is listed as a threatened species in New South Wales (Vulnerable: Biodiversity Conservation Act 2016). Squirrel gliders prefer dry sclerophyll forests, with populations found in remnant bushland along the highly urbanised east coast of Australia and the Great Dividing Range (Pavlova et al. 2010). In addition to their reliance on tree hollows, more than 50% of their diet depends on the nectar, pollen and sap of five plant species (belonging to the Eucalyptus, Corymbia, Melaleuca, Banksia genera) (Ball et al., 2009). Eucalyptus forests have experienced the greatest deforestation of Australian vegetation and 80% of remnants have been modified by human activity (Bradshaw 2012). Habitat loss and fragmentation are the main threats to squirrel gliders as described in the Action Plan for Australian Mammals (Goldingay and Sharpe 2004; Woinarski et al. 2014). The fragmentation of forests can lead to a loss of squirrel glider genetic diversity through genetic drift (Frankham et al. 2002) while also increasing intraspecific competition for tree hollows and other habitat resources.
Squirrel gliders rely on trees for dispersal, and as such gene flow between populations can be obstructed by gaps in canopy that exceed their maximum glide distance of 70 m or by short trees that do not support a glide angle of 28.5 degrees (Van der Ree, 2002; Van Der Ree et al., 2004; Goldingay and Taylor, 2009). Additionally, squirrel gliders generally avoid remnant trees in the urban matrix as they are sensitive to light pollution, road density and associated noise (Francis et al., 2015). Limited access to food, as a result of land clearing and fragmentation, reduces the effective population size of squirrel gliders (Sharpe 2004). Evidence suggests that small isolated patches must be linked to a larger metapopulation to prevent inbreeding depression and localised extinction (Goldingay and Sharpe, 2004; Van Der Ree et al., 2004). Many studies have relied on radio-tracking to infer connectivity of patches and dispersal of squirrel gliders (van der Ree and Bennett, 2003; Sharpe and Goldingay, 2007; Goldingay et al., 2010; Brearley et al., 2011; Soanes et al., 2015), however few have examined genetic structure and gene flow to make this inference.
The emergence of landscape genetics (Manel et al. 2003) contributed substantially to the field of ecology, evolution, and species conservation in recent decades. Among others, landscape genetics combines population genetics, landscape ecology and spatial analytical techniques to quantify the effects of landscape composition, configuration, and matrix quality on gene flow using neutral genetic data (Balkenhol et al. 2015). The field has rapidly moved from the more traditional isolation-by-distance analyses to explain differences in the genetic structure of populations that drill down further into movement and dispersion dynamics. Estimates of gene flow can be tested against measures of effective geographic distance to find estimates of landscape resistance that best fit with the genetic data. Landscape genetics has proven useful to quantify the effects of habitat fragmentation on genetic structure, and identify landscape features that are important for maintaining genetic connectivity between populations within heterogeneous landscapes (e.g. Sharma et al. 2013), as well as pinpoint barriers to gene flow (Cushman and Lewis 2010).
Large-scale studies have compared the genetic diversity of squirrel gliders across the entire east coast of Australia (Pavlova et al. 2010; Taylor et al. 2011), but key squirrel glider hotspots remain unsampled. Additionally and to our knowledge, only one published study has examined the effect of fragmentation on a fine-scale, in Mackay and Brisbane (Queensland) (Goldingay et al. 2013). Assessing the fine-scale genetic structure of squirrel glider populations would assist with the development and use of local conservation management strategies in areas where they are threatened. Thus far, squirrel glider research has only used mitochondrial (cytochrome b) and microsatellite DNA markers to examine genetic structure (Pavlova et al. 2010; Taylor et al. 2011; Goldingay et al. 2013; Dudaniec et al. 2016), and researchers have yet to take advantage of the power of next-generation sequencing technology and single nucleotide polymorphisms (SNPs).
SNPs are the most common form of sequence variation, characterised by single base nucleotide changes in the genome that differ among individuals (Brumfield et al. 2003). Advances in next-generation sequencing means that tens of thousands of bi-allelic, co-dominant SNP markers can be detected in the genome (Kumar et al. 2012). SNPs have wider genome coverage than microsatellites such that population samples of four individuals are enough to identify population structure (Liu et al. 2005; Shi et al. 2010; Lah et al. 2016; Dussex et al. 2018). Genome-wide SNP discovery with next-generation sequencing is gaining importance in conservation genetic research on threatened species given that small sample sizes are common. Additionally, genome-wide SNPs hold an advantage over microsatellites when it comes to inferring demographic history and genetic diversity estimates for conservation management purposes (Zimmerman et al. 2020). Genome-wide SNPs have evaluated the success of threatened Australian marsupial translocation programs in recent years, including that of the greater bilby, Macrotis lagotis and western barred bandicoot, Perameles bougainville (White et al. 2018). Furthermore, SNPs have effectively informed conservation management of threatened Australian marsupials including the koala, Phascolarctos cinereus (Kjeldsen et al. 2016) and the Tasmanian devil, Sarcophilus harrisii (Wright et al. 2019). SNPs should therefore be extremely valuable in assessing the effects of habitat fragmentation on squirrel glider populations at a fine-scale.
Lake Macquarie Local Government Area (LGA) is a nationally significant area within the distribution of squirrel gliders since it is believed to contain the highest density of the species across their range in the eastern seaboard of Australia (estimated 5000 individuals in the Lake Macquarie – Wyong area, concluded from the Wildlife Atlas records in New South Wales) (Smith 2002; Fallding 2015). Because of this, it is essential to monitor squirrel gliders in the area to ensure habitat fragmentation and biogeographical barriers do not negatively impact upon the conservation status further. Here we use mitochondrial DNA and genome-wide SNP markers to examine the demographic history and effect of habitat fragmentation on the population structure of threatened squirrel gliders in the Lake Macquarie LGA. We hypothesised that: (1) squirrel gliders in isolated patches would have low levels of genetic diversity and limited gene flow; (2) squirrel glider populations isolated by biogeographical barriers (Lake Macquarie and the Pacific Ocean) would be highly differentiated from other populations; and (3) landscape heterogeneity (vegetation and land use) would impact the genetic structure and differentiation of squirrel glider populations. This is the first study to report on the genetic structure of the Lake Macquarie LGA squirrel glider metapopulation, despite the LGA being a stronghold population for the species (Smith 2002; Fallding 2015).
Methods
Study area
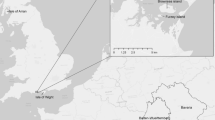
Our study was undertaken in the Lake Macquarie LGA, ~ 130 km north of Sydney, Australia (Fig. 1). Lake Macquarie LGA has experienced a high level of land clearing resulting in fragmentation of forests that were recognised as critical squirrel glider habitat (Smith 2002; Fallding 2015). In addition to habitat fragmentation, squirrel gliders also face added pressure from biogeographical barriers in the landscape, namely a large natural saltwater lake (Lake Macquarie, 120 km2) that presents a barrier to gene flow in the LGA as well as the Pacific Ocean on the east coast (Fig. 1).

Squirrel glider trapping locations from 2017–2020. Black dots illustrate locations where squirrel gliders were caught. Location codes: WYB = Wyee Road (Nth), WYC = Wyee Road (Sth), WR = Wilton Road (Est), SP = Sunshine Park, OR = Overhill Road, RA = Ruston Avenue, CROB = Croudace Bay, GMP = George McGregor Park, FERNS = Fernleigh Track (Sth), OCN = Ocean Street, FERNN = Fernleigh Track (Nth), DUD = Dudley Beach Road, RHL = Red Head Lagoon, GSCA = Glenrock SCA. The 30-meter-wide powerline easement is located at WR
We selected squirrel glider trapping locations in the Lake Macquarie LGA through a combination of spatial modelling and visual identification of suitable vegetation. Sites required dens (large hollow bearing trees) and eucalypts (Corymbia maculata, Eucalyptus robusta, Eucalyptus tereticornis), acacias (Acacia irrorata) and banksias (Banksia spinulosa; Banksia integrifolia) since squirrel gliders are known to feed on their sap, gum and nectar (Ball et al. 2009). Additionally, Smith and Murray (2003) found higher densities of squirrel gliders in the Lake Macquarie area associated with an understory of Xanthorrhoea and Banksia species, and scribbly gum (Eucalyptus haemastoma or racemosa), smooth-barked apple (Angophora costata) and red bloodwood trees (Corymbia gummifera). Once potential sites were located, we referred to BioNet records (NSW Office of Environment and Heritage 2021), and used spotlighting surveys and/or infrared cameras (Gracanin et al. 2019) to confirm glider presence at a site prior to live trapping.
Sample collection and DNA extraction
We conducted live trapping at 32 locations over a four-year period, with squirrel gliders captured at 14 locations (Fig. 1). Each site was subject to at least one week of live trapping using 12 traps per site (two rows of six with each trap spaced 50 m apart), and further focus was given to sites that captured squirrel gliders during this period to increase sample size. Once squirrel gliders were detected at a site, we re-trapped the area every six months to a year across the survey period. We used a combination of Mawbey traps, Elliot B traps, cage traps and Winning and King pipe traps to live trap gliders (Mawbey 1989; Quin 1995; Winning and King 2008). All traps were secured to trees so that the trap entrance was at least two meters from ground height. Each trap contained a bait ball made from a mixture of peanut butter, honey, and oats (Winning and King 2008), leaves for insulation, and we sprayed a 1:4 ratio of honey:water around each trap entrance and up the tree to a height of 6 m as a further attractant (Sharpe and Goldingay 2007).
Traps were checked each morning at sunrise. We weighed, sexed, measured (tail length, right hind foot length, head width and head length) and marked each squirrel glider with a metal ear tag or an ear punch combination. Before releasing the squirrel gliders, we collected DNA samples from 94 squirrel gliders in the form of ear tissue (or in the early stages of the project, a buccal swab, n = 30), using a 2 mm metal ear punch (Able Scientific, Australia) that was previously sprayed with 70% ethanol and flamed for sterilization. Once cooled, a 2 mm tissue punch was taken from the outside edge of the ear and stored in sterilized vials containing 80–95% ethanol. DNA was kept at – 20 °C prior to DNA extraction. The processing of each squirrel glider was limited to a maximum time of five-minutes, and individuals were released on the same tree in which they were caught.
We extracted genomic DNA from the buccal swab and ear tissue samples using the One-4-ALL Genomic Miniprep Kit (BioBasic Inc. Ontario, Canada) and DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions.
Mitochondrial DNA: cytochrome b gene
We used genomic DNA samples extracted from buccal swabs and ear tissue for mitochondrial DNA analysis. We amplified a 385 base pair fragment of the mtDNA cytochrome b gene using primer pair L14724 (5’-CGA AGC TTG ATA TGA AAA ACC ATC GTT G-3’) and H15149 (5’-AAA CTG CAG CCC CTC AGA ATG ATA TTT GTC CTC A-3’) as described by Kocher et al. (1989) and Irwin et al. (1991). PCR products were treated in the post-PCR lab via the ExoSAP-IT™ enzymatic method (Affymetrix Inc. USB Corp., Cleveland, Ohio) prior to sequencing with an ABI PRISM BigDyeTM Terminator Cycle Sequencing Kit (Applied Biosystems, USA). We cleaned the sequencing products with a precipitation cleanup method and sequences were generated with a 3130xl Genetic Analyzer (Applied Biosystems, Australia), at the University of Wollongong, School of Biological Sciences. We visually inspected and manually edited the sequences using the program ChromasPro v 1.33 (Technelysium Pty Ltd, Brisbane) and aligned them using BioEdit v 7.2.6.1 (Hall 1999). Cytochrome b sequences were obtained from 55 squirrel glider samples in the Lake Macquarie LGA, and from 15 buccal swab samples that were donated to the project from the Forster region, approximately 130 km northeast of Lake Macquarie. Therefore, cytochrome b sequences were generated for a total of 70 individual squirrel gliders (Table 1).
All mtDNA haplotypes were identified as a unique arrangement of nucleotides in the cytochrome b DNA sequence of individuals. Haplotype diversity and nucleotide diversity were calculated for the Lake Macquarie and Forster regions. Haplotype diversity (h) gave the probability that two randomly sampled alleles would be different (probability 0–1), while nucleotide diversity (π) determined whether the differences between haplotypes were minor (approaching zero) or major (approaching 1). Tajima’s D and Fu’s Fs tests were used to test the neutrality of the mitochondrial DNA sequences within populations and regions (Tajima 1989; Fu 1997), and a mismatch distribution analysis was conducted to determine whether squirrel gliders had undergone a recent range expansion (Schneider and Excoffier 1999). All of these metrics were calculated in DnaSP v 5.10 (Librado and Rozas 2009). A median-joining network was constructed in PopART to visualise relationships among haplotypes (Bandelt et al. 1999).
The mutation rate of the cytochrome b gene was calculated to gain insight into the demographic history of squirrel gliders. To do this we trimmed 70 cytochrome b sequences and aligned them to closely related species. This included a 374 bp cytochrome b sequence from a sugar glider (Petaurus breviceps, GenBank Accession Number FJ657661.1), a mahogany glider (Petaurus gracilis, GenBank Accession Number HQ284045.1) and a leadbeaters possum (Gymnobelideus leadbeateri, GenBank Accession Number NC_053898.1). The alignment was imported to BEAUti v2.6.6 to create a file for BEAST analysis (Bouckaert et al. 2019). We selected a General Time Reversible (maximum-likelihood GTR) site model, with a strict molecular clock and calibrated yule model for priors.
In priors, sugar gliders and squirrel gliders were combined as most recent common ancestors, and monophyly was enforced. To calibrate the tree, we selected a Normal distribution, and the mean was centered at 1.73 million years with a standard deviation of 0.5 million years (30% of the divergence estimate). This mean was taken from Meredith et al. (2010) after examining genetic research and fossil information provided by Malekian et al. (2010) and Meredith et al. (2010). Finally, all taxa were combined as most recent common ancestors and monophyly was enforced. The mean was centered at 15.97 million years before present as that’s when the Leadbeaters possum is estimated to have diverged from gliders (Meredith et al. 2010). A standard deviation of 5 million years was selected (30% of the divergence estimate). The parameters for the molecular clock rate (clockRate) and the speciation rate (birthRateY) of the Yule tree prior were entered as Alpha = 0.001 and Beta = 1000. Markov chain Monte Carlo runs were set to 10,000,000 generations. The output file was generated and run through BEAST v2.6.6 (Bouckaert et al. 2019). The resulting tree was visualized in FigTree v1.4.4 (Rambaut 2019) and the log file was analyzed in Tracer v1.7.2 to estimate the mutation rate of the cytochrome b gene in squirrel gliders (Rambaut et al. 2018). We used this mutation rate to estimate the average number of generations taken for a single mutation to be acquired, assuming a generation time of 4.5 years as per Pavlova et al. (2010).
Finally, we used the mutation rate from the previous analysis to estimate the female effective population size of squirrel gliders in the Lake Macquarie LGA. We displayed the female effective population size with a Bayesian skyline plot in BEAST as per Drummond et al. (2005). Changes to the female effective population size were examined through time.
Nuclear DNA: SNPs
We examined squirrel glider genomic DNA samples on 1% agarose gel electrophoresis and with a NanoDrop 2000 Spectrophotometer (Thermo Scientific) to identify samples with appropriate concentration and purity for next-generation sequencing. The buccal swab genomic DNA samples did not pass this quality control stage. Therefore, we selected 94 tissue DNA samples (belonging to 89 squirrel glider individuals) for sequencing.
We plated and sent genomic DNA to Diversity Arrays Technology (DArTseq), University of Canberra, Australia, for concentration and sequencing (Kilian et al. 2012). DArTseq is a high throughput method that combines genome complexity reduction methods and next-generation sequencing to detect a large number of SNPs at a relatively low cost (Kilian et al. 2012). Additionally, DArTseq optimises the technology for each species by selecting the most appropriate complexity reduction method. For this squirrel glider study, the PstI-SphI method was used prior to PCR amplification and sequencing of fragments on an Illumina Hiseq2500. This PstI-SphI restriction enzyme combination has proved extremely efficient in detecting SNPs in Australian marsupials (Schultz et al. 2018; Kjeldsen et al. 2019; Wright et al. 2019), including sugar gliders (Petaurus breviceps) (Knipler et al. 2021).
DArTseq aligned the squirrel glider sequences to the Leadbeaters possum (Gymnobelideus leadbeateri) reference genome and retained SNPs with a minimum sequence identity of 70%. Calling quality was assured with a high average read depth per locus (over 20 reads per locus average across all markers). Following this, we filtered SNPs and individuals using dartR 1.1.11 (Gruber et al. 2018) in R 4.0.2 (R Core Team 2015). Duplicate samples of lower quality were removed from the dataset so that only unique individuals remained. Loci were removed if the call rate was < 0.95, if they had reproducibility < 0.99, and if the hamming distance was < 0.2. Loci were also removed if they deviated from Hardy-Weinberg Equilibrium (p < 0.05) and the SNP data set was analysed to ensure no monomorphic loci remained. The resulting dataset was used for inferring demographic history as it contained linked loci (8,580 SNPs retained). We used TASSEL v5.2.78 (Bradbury et al. 2007) to test for neutrality in the form of Tajima’s D (± SD) using a step size of 100 and a window size of 500 for populations with ≥ 4 samples. Current effective population sizes (Ne) were calculated for each population using the larger partially filtered dataset of SNPs and NeESTIMATOR v2.1 (Do et al. 2014). A critical threshold value of 0.05 was selected and random mating was assumed. After this, we further filtered the dataset to remove linked loci using the “gl.filter.secondaries” function in the dartR package. We conducted an OutFLANK outlier test in R to infer the neutrality of SNPs (Whitlock and Lotterhos 2015). No loci were identified as being under selection, so all were retained, and this filtered dataset was used for further population genomic analyses (6,834 SNPs).
We undertook all subsequent analyses in R 4.0.2 (R Core Team 2015) unless otherwise stated. We calculated observed and expected heterozygosity for each location to gain insight into the genetic diversity of squirrel glider populations. Observed heterozygosity (Hobs) was calculated with the R package dartR, while expected heterozygosity (Hex) was calculated with the package adegenet 2.1.3 (Jombart 2008). We also calculated the inbreeding coefficient for each squirrel glider location (FIS = 1-Hobs/Hex), and the overall average FIS for the total region. The nucleotide diversity of SNPs (π ± SD) was calculated for each population in TASSEL as a measure of genetic variation.
We examined structure and population differentiation among squirrel gliders in Lake Macquarie LGA using Analyses of Molecular Variance (AMOVA’s), and we plotted a Principal Coordinate Analysis (PCoA) to assist with the visualisation of this genetic structure. The PCoA was conducted in dartR. We ran the AMOVA’s with the R package poppr 2.9.0 using 9,999 permutations (Zhian Kamvar 2021). The first AMOVA examined the genetic differentiation of squirrel gliders in two regions: west (locations WYB, WYC, WR, SP, OR) and east (locations RA, CROB, GMP, FERNS, OCN, FERNN, DUD, RHL, GSCA) of Lake Macquarie. The second AMOVA examined the genetic differentiation of squirrel gliders across all 14 populations, and the third served as a control with two haphazardly drawn lines dividing the sampling locations into three groups (group 1: WYB, WYC, SP, group 2: WR, OR, CROB, RA, FERNS, OCN, and group 3: GMP, FERNN, DUD, RHL, GSCA). Next, we examined genetic differentiation in the form of pairwise FST between the 14 locations of squirrel gliders. Pairwise FST values were generated with the package dartR and 10,000 bootstraps were performed across loci to generate the corresponding p-values. Similarly, the average FST was calculated for the overall region.
We further examined squirrel glider genetic structure with STRUCTURE 2.3.4 (Pritchard et al. 2000). STRUCTURE uses a Bayesian clustering approach to assign individuals to groups (“K”) where members display similar patterns of genetic variation. These groups are indicative of ancestral populations that individuals may have originated from (Pritchard et al. 2000; Pritchard et al. 2003). We ran the analysis eight times for each K value (1–15) with a 10,000-length burn-in period and 10,000 Monte Carlo Markov Chain replications. The most likely K values were selected after viewing the results in Structure Harvester (Web v0.6.94) and referring to the Evanno method and the largest Delta K value (Evanno et al. 2005; Earl and VonHoldt 2012).
Landscape genomic analyses
In addition to identifying genetic differentiation and genetic structure, we wanted to identify landscape features that influenced (diverting, preventing, or enhancing) squirrel glider gene flow in areas of the Lake Macquarie LGA. Genomic data was combined with spatially-explicit data to investigate the effect of geographic distance and habitat heterogeneity on genetic distance of squirrel glider populations. First, we undertook an isolation-by-distance (IBD) analysis in the dartR package using 9,999 permutations. This mantel test took the log of the straight-line Euclidean distances between pairwise populations and compared them to the genetic distance matrix of populations in the form of FST/(1-FST), to see if there was a significant relationship between geographic distance and genetic distance of populations. The pairwise FST values from dartR were used in this case. We used the results of this IBD analysis as a baseline, to run least cost path analyses and determine whether they could explain more of the genetic variation in the landscape.
We ran two separate least cost path (LCP) analyses using the “gl.genleastcost” function in the dartR package. LCP approach estimates the shortest distance between target core areas while accounting for resistance to movement (inferred here from the genetic data) (Adriaensen et al. 2003). LCP requires an input resistance surface, also called a friction matrix, and that is a spatial layer reflecting the degree to which a location in the landscape facilitates or impedes movement of a focal species (Zeller et al. 2012).
We created two friction matrices in ARCMAP 10.7.1 (ESRI, California) for each LCP scenario. The first friction matrix only considered water bodies as biogeographical barriers. Hence, the Lake Macquarie and the Pacific Ocean were manually assigned “NoData” cell values, while all terrestrial areas were given a cell value of 1, assuming constant movement capacity across a homogeneous terrestrial landscape within our study area. This analysis will hereafter be referred to as an isolation by barrier analysis.
The second friction matrix used the Plant Community Type (PCT) spatial layers from Bell et al. (2016) and Eco Logical Australia Pty Ltd (2003), and land use spatial layers from the NSW Government (2007). We assigned values to the cells (pixels in the map) based on our opinion, the literature, and a range of test models we ran. Values scaled from 1 for optimal habitat with low resistance that eased movement, to 200 for unsuitable habitat with high resistance that would render movement (and hence dispersal) challenging. We divided vegetation layers into five categories based on the diet and habitat requirements of squirrel gliders (Smith 2002; Smith and Murray 2003; Ball et al. 2009; Niche Environment and Heritage 2013; Payne 2016): highly suitable vegetation (1), suitable vegetation (10), moderately suitable vegetation (20), moderately unsuitable vegetation (75) and unsuitable vegetation (100). Appendix 1 details how the 62 PCTs were assigned. Land use was divided into the following classes: Urban residential (80), roads (100), railways (80) (because there is less traffic and the width is smaller than roads), rural residential without agriculture (70), rural residential with agriculture (100), native/exotic pasture matrix (100), grazing irrigated modified pastures (100), irrigated turf farming (100), mines/quarries and large cleared areas (200), the Lake and Ocean (NoData, aka complete barrier to dispersal). Highly suitable vegetation was given a 35-meter buffer so that two patches either side of a barrier (e.g. road/river) would intersect within maximum gliding distance (70 m (Van der Ree and Bennett 2003)), indicating crossing potential. If cells overlapped between vegetation and land use, then the minimum value was selected for the final friction matrix. This analysis will be heron referred to as a least cost path analysis.
We computed new geographic distances using the two friction matrixes (isolation by barrier raster and the least cost path raster, cell factor = 15, function = mean in ArcGIS) and the spatial coordinates of individuals. These new pairwise geographic distances were then tested for correlation with genetic distances and number of neighbours = 8 with the “gl.genleastcost” function of the R package dartR. We then compared the statistics from each spatial analysis (IBD, isolation by barrier analysis and least cost path analysis) to find the best fit (Milanesi et al. 2016).
Results
Trapping
We trapped squirrel gliders in 14 out of the 32 sites surveyed. We collected ear tissue samples from 94 squirrel glider individuals, while an additional 30 squirrel gliders had buccal swab samples collected. We caught many male and female squirrel gliders over consecutive years in the original location they were trapped, suggesting site fidelity. For example, we recaught a male and female squirrel glider after two years within 50 m of their original detection at location RA, while a male squirrel glider was recaught after two years within 100 m of its original detection at location GSCA. Additionally, we caught one adult male and one adult female squirrel glider on either side of a 30-meter powerline easement during one week of live trapping (location WR).
Cytochrome b
Of the 70 squirrel gliders sequenced, nine polymorphic sites were identified (2.34%), 376 conserved sites (97.66%) and eight haplotypes. The average total nucleotide diversity was low for both Lake Macquarie LGA and Forster (Lake Macquarie π = 0.002 ± SD 0.000, Forster π = 0.002 ± SD 0.003) and total haplotype diversity was moderately high for Lake Macquarie LGA (h = 0.648 ± SD 0.052) but lower in Forster (h = 0.448, ± SD 0.134). A BLASTn search of GenBank failed to find a match for haplotypes H02 (habitat fragment MP, refer Table 1), H04 (locations RHL, GSCA, OCN, SP, WYB) and H07 (habitat fragment MP, refer Table 1), therefore these haplotypes are believed to be unique to this study. Haplotype three (H03) was the most common, occurring in 57.14% of individuals and most of the locations (Table 1). In comparison, haplotype two (H02) was only observed once and it was unique to Lake Macquarie’s MP location. Haplotype seven (H07) was also unique to the MP location at Lake Macquarie. Locations SP and OR had a single haplotype at each site (Table 1). Lake Macquarie Tajima’s D and Fu’s F values indicated population expansion and an excess of rare mutations in the Lake Macquarie squirrel glider populations; however, these values were not significant (-1.07922 and − 2.254 respectively, p > 0.10) (Table 1). Similarly, the results of the mismatch distribution analysis produced a unimodal curve. The overall average number of pairwise differences was k = 0.951 and the raggedness statistic was r = 0.0588. The mutation rate for the squirrel glider cytochrome b gene was calculated to be 1.63% per site per million years. The estimated time for a single mutation was 164,036 years (lower 95% HD = 312,407, upper 95% HD = 102,053), or 2.74329E-05 mutations per generation (Fig. 2A), and a star-like haplotype network was produced (Fig. 2B). The Bayesian skyline plot depicted a constant female effective population size over the last 200 years (Appendix 2).

(A) Maximum-likelihood tree reflecting divergence times of cytochrome b haplotypes in leadbeaters possum (G. leadbeateri, n = 1), mahogany glider (P. gracilis, n = 1), sugar glider (P. breviceps, n = 1) and squirrel glider (P. norfolcensis, n = 70). Timeline in millions of years before present (MYA). (B) MtDNA median-joining network displaying the relationship among eight cytochrome b haplotypes (H01-H08) of squirrel gliders in Lake Macquarie and Forster, NSW (n = 70). A slash in the branches linking two haplotypes indicate one nucleotide substitution and two slashes indicate two nucleotide substitutions. Refer to Table 1 to see where each haplotype was found
Genetic differentiation and structure (SNPs)
DArTseq returned 22,579 loci from 92 squirrel glider samples and 14 locations in the Lake Macquarie LGA. Two squirrel glider samples did not meet quality control standards and were excluded. During the final filtering stage in R, five duplicate squirrel glider samples were removed. Two datasets were generated after stringent filtering: one for inferring demographic history as it contained linked loci (8,580 loci, 87 squirrel glider individuals, 14 locations in the Lake Macquarie LGA), and one for subsequent population genetic analyses (6,834 loci, 87 squirrel glider individuals, 14 locations in the Lake Macquarie LGA).
Tajima’s D was calculated for populations with ≥ 4 samples using the partially filtered dataset that contained linked SNPs. All values were negative, which is indicative of population expansion after a recent genetic bottleneck (Fig. 3). Care should be given when interpreting these results due to small sample sizes (n = 4–16) (Subramanian 2016). Effective population sizes of squirrel gliders in Lake Macquarie populations were very small (Table 2) and should be viewed with caution due to the small sample sizes. These values should be used as a comparison between populations to identify regions of concern, rather than an accurate estimate of effective population size (Zachos et al. 2016).

Box-plot of Tajima’s D statistics for locations with ≥ 4 squirrel glider samples in Lake Macquarie LGA (8,580 loci, 87 individuals)
Using the strictly filtered SNP dataset of 6,834 loci, we compared observed heterozygosity to expected heterozygosity at each location, to understand the levels of squirrel glider genetic diversity. Observed heterozygosity was higher than expected heterozygosity at every location except for location WR, however the values were low indicating low levels of genetic variability (Table 2). There was no evidence of inbreeding (overall FIS = 0.015). The nucleotide diversity of the SNPs displayed similar patterns to the mtDNA results, with population OR, GMP and OCN having low nucleotide diversity. Three AMOVA’s examined the genetic variation of squirrel glider populations. Although west and east Lake Macquarie explained 7.79% of the genetic variation, this value resembled the variation observed between the random control groups (5.51%) (Table 3). When the 14 sampling locations (i.e. putative populations) were included in an AMOVA, further genetic variation was explained (15.12%) (Table 3) (Fig. 4). The cumulative percentage of genetic variation explained by the first four axes was 23.2%, with axis 1 accounting for 8.6% of the variation (Fig. 4). The most important observations were that the southwestern squirrel glider populations (OR, SP, WR, WYB, WYC) separate out from the northeastern populations on axis 1, while FERNN and OCN each appear to separate out on axis 2. Axis 4 depicts a clear separation of population OR.

Graphical representation of the Principal Coordinates Analysis (PCoA) based on the genetic distances of squirrel gliders from 14 putative populations (“pop”) and 6,834 loci. (A) The first two PCoA axes explain 14.4% of the observed genetic variation when combined. (B) The third and fourth PCoA axes explain 8.8% of the observed genetic variation when combined. N = 87
We compared genetic differentiation of squirrel glider populations through pairwise FST. Statistically significant pairwise FST values ranged from 0.014 (DUD vs. RHL) to 0.335 (CROB vs. SP) (Table 4). FST values greater than 0.15 are indicative of significant differentiation and genetic structure (Frankham et al. 2002), and in this case many of the pairwise comparisons produced results that indicated considerable differentiation (Table 4). Squirrel gliders at location OR were greatly differentiated from all other areas (FST 0.149 to 0.273), and CROB, FERNN, OCN and SP displayed substantial genetic differentiation from most other areas (Table 4). As a result, the average FST was quite high with a value of 0.148.
STRUCTURE analyses provided further insight into the genetic structure of squirrel gliders in the Lake Macquarie LGA. The Structure Harvester results showed that Delta K had a peak at K = 2 (Delta K = 47.57), with a small peak again at K = 6 (Delta K = 5.22) (Appendix 3). Results for both are presented here since there can be bias towards finding K = 2 with the Delta K method (Cullingham et al. 2020). Upon visualising the K = 2 STRUCTURE admixture plot, it was clear that the squirrel glider individuals were divided into two clusters, similar to the results on axis 1 of the PCoA plot (populations northeast of the lake and populations southwest of the lake) (Fig. 5). When K = 6, unique clusters appeared within the FERNN, OCN, OR, and WR populations, suggesting that the Lake Macquarie individuals could have been derived from six squirrel glider ancestral sources (Fig. 5). The separation of these populations (FERNN, OCN, OR and WR) reflects the patterns observed in the PCoA plots (Figs. 4 and 5). Admixture was present in all sampling locations; however, some individuals were pure to clusters within FERNN, GSCA, OCN, OR and WR (q value > 0.8).

STRUCTURE admixture results for 87 squirrel glider individuals, 6,834 loci and K = 2 (above) and K = 6 clusters (below). Each individual is represented by a vertical bar while putative populations are divided by the white dashed lines and labelled below the plot. Each cluster is displayed with a unique colour
Spatial analyses
With the isolation by distance analysis, we found a significant relationship between geographic straight-line distances and genetic distances of squirrel glider populations (R2 = 0.3227, p < 0.01) (Table 5). Second, an isolation by barrier analysis was performed to see if the biogeographic barriers explained more of the genetic variation (Lake Macquarie and the Pacific Ocean). The R2 value did improve (R2 = 0.5390, p < 0.001) (Table 5), but it did not consider habitat heterogeneity. Thirdly, we ran a least cost path analysis to include landscape features and habitat heterogeneity (biogeographical barriers, vegetation, and land use). When run, it was clear that the least cost paths between populations (Fig. 6) explained the greatest proportion of genetic variation between populations (R2 = 0.6467, p < 0.001) (Table 5).

Friction matrix and least cost paths between 14 squirrel glider populations in Lake Macquarie. Raster was generated in ARCMAP and the least cost paths were created with the R package dartR. Cost raster cell values ranged from optimal habitat (1) to unsuitable habitat (200) based on vegetation communities and land use type. The lake and ocean were given “NoData” values and assumed a complete barrier to dispersal, except where the vegetation buffer allowed crossing using maximum gliding distance of 70 m
Discussion
Woinarski et al. (2014) lists habitat fragmentation as the main threat to squirrel gliders, which is particularly concerning in the Central Coast of NSW (Smith 2002; Fallding 2015). In this study, we examined whether the effects of habitat fragmentation were detectable on the populations genetic structure. We expected that squirrel gliders in isolated patches would have low levels of genetic diversity and limited gene flow. We also sought to identify areas that would enhance gene flow between populations in our study area and identify areas that present potential barriers to dispersal.
Our first hypothesis was partly rejected as we detected high haplotype diversity in squirrel glider populations, despite previous research recording low haplotype diversity in squirrel glider populations 40 km from Lake Macquarie in Port Stephens (h = 0.18), and 20 km from Lake Macquarie in Warnervale (h = 0.50) (Pavlova et al. 2010). There did not appear to be haplotypes grouped with respect to any geographical barrier or feature. MtDNA has a relatively fast mutation rate and is useful for determining the genetic history of populations (Arif et al. 2011). High haplotype diversity and low nucleotide diversity in the Lake Macquarie squirrel glider population points to a historical genetic bottleneck followed by rapid expansion and recovery from a small effective population size (Grant and Bowen 1998; Avise 2000). This is reflected in the star-like haplotype network, which is consistent with what is displayed after a historical genetic bottleneck with many haplotypes branching from the dominant central H03 haplotype (high haplotype diversity) depicting minimal nucleotide differences (low nucleotide diversity). The unimodal curve from the mismatch distribution and negative Tajima’s D values for both the mtDNA sequences and the SNP dataset reinforce this concept (Tajima 1989). It was estimated to take 164,036 years for a mutation to occur in the cytochrome b gene. This information paired with the star-like haplotype network indicates that a range expansion occurred in the mid-late Pleistocene, which supports the results of Pavlova et al. (2010).
We also used SNPs to examine whether the genetic diversity of squirrel gliders would be low in habitat patches within Lake Macquarie LGA. Low genetic diversity has been observed in arboreal mammal populations within fragmented landscapes such as the Siberian flying squirrel (Pteromys volans) (Lampila et al. 2009) and ringtail possum (Pseudocheirus peregrinus) (Lancaster et al. 2016). Despite these predictions, the genetic diversity values did not differ greatly between the mtDNA and SNP data, and squirrel gliders displayed higher than expected levels of observed heterozygosity within the Lake Macquarie LGA, with no evidence of inbreeding (FIS = 0.015). Higher than expected heterozygosity could potentially be explained by the genetic bottleneck detected with the mtDNA analyses, as bottlenecks lead to a quick loss in rare alleles while heterozygosity decreases slowly over time (Lampila et al. 2009). Additionally, excess heterozygosity in current squirrel glider populations could be the result of admixture and population growth following the historic bottleneck. This concept is supported by the mtDNA analyses and the nuclear FST and STRUCTURE results. Taken together, the data suggest that the impacts of urban fragmentation of the Lake Macquarie forest system has not yet produced a substantial loss of heterozygosity with the associated risks of loss of fitness due to inbreeding effects (Frankham et al. 2002). Nevertheless, restriction of squirrel gliders to smaller and more isolated habitat fragments is likely to produce a drastic effect in coming generations. Therefore, we recommend continued monitoring of heterozygosity in isolated populations.
Genetic differentiation (i.e. structure) between fragments was expected to be quite high due to isolation by both the lake and extensive urbanisation. This expectation was based on the fact that squirrel gliders are sensitive to noise pollution in the urban matrix (Francis et al. 2015) and have a maximum glide distance of 70 m (Van der Ree and Bennett 2003). This theory was supported with evidence of high levels of genetic structuring in the Lake Macquarie squirrel glider populations. The FST values were unusually high considering the fine scale of this study, and they were considerably higher than sugar glider populations in the same study area (FST values ranging from 0.011 to 0.131 based on 11,292 SNPs) (Knipler et al. unpublished data). Despite the two species sharing similar life history traits and requiring similar habitat (Lindenmayer 2002), the difference was considerable (squirrel glider FST values from 0.014 to 0.335). For example, we found a high level of genetic differentiation between squirrel gliders at locations OR and WR (FST = 0.149), but in Knipler et al. (unpublished data) the FST value for sugar gliders between OR and WR was significantly lower (FST = 0.042). Squirrel gliders had greater pairwise FST values for every location where squirrel gliders and sugar gliders were present together (OR, SP, WR, WYB, WYC) (Knipler et al., unpublished data). It is possible that squirrel gliders are more specialised and sensitive to the urban matrix and that is why they are experiencing higher levels of differentiation than sugar gliders. This warrants further investigation as population differentiation is characteristic of most specialist, arboreal mammals in fragmented habitats (Barratt et al. 1999; Larsen et al. 2013; Baden et al. 2014; Sgarlata et al. 2016).
While there was an isolation-by-distance pattern of gene flow in the squirrel glider populations in the Lake Macquarie LGA, it did not explain a large proportion of the genetic variation. Microsatellite analyses by Goldingay et al. (2013) for squirrel glider populations in the cities of Mackay and Brisbane, Queensland, found no isolation-by-distance effect, with strong genetic divergence occurring in squirrel glider populations 3 km apart after 30 years of landscape change. Goldingay et al. (2013) suggests that genetic drift in habitat fragments can have a stronger effect on allele frequencies than geographic distance. In the Lake Macquarie LGA, there was evidence of significant genetic differentiation between squirrel glider locations that were geographically close to each other. For example, we detected high levels of genetic differentiation between two locations that were only 1.8 km apart but divided by urban development in the town Whitebridge (FERNN and OCN FST = 0.179), and locations 900 m apart but divided by urban development in the town Valentine (CROB and RA FST = 0.131). This contrasts with squirrel glider samples that were collected 2.2 km and 1.5 km from each other but connected by forest (WYB and WYC FST = 0.087, DUD and GSCA FST = 0.027). This is where the landscape genetic analyses proved useful. By combining next-generation sequencing and landscape genetic analyses our study was able to explain a large portion of the genetic variation observed in the squirrel glider populations. To our knowledge, just one other study has used resistance models and least cost path analyses to infer genetic structure of squirrel gliders in the past (Dudaniec et al. 2016), but the proportion of variation explained by their model was lower than ours (R2 = 0.027 versus R2 = 0.647). A possible reason for this improvement is the incorporation of genome-wide SNPs rather than microsatellites, and the use of a 35-meter buffer to account for the maximum glide distance of 70 m (Van der Ree and Bennett 2003).
Following the least cost path analysis, we identified thin vegetation strips as possible corridors to enhance squirrel glider gene flow in the following locations: Fishery Point Road (Bonnells Bay), Kilaben Bay (Rathmines), Fernleigh Track (Whitebridge), vegetation west of Fernleigh Tack (Charlestown), Pacific Highway crossing (Jewells to Bennetts Green), vegetation patches between Glenrock State Conservation Area (Highfields) and Blackbutt Nature Reserve (New Lambton), and vegetation along creek lines (Fig. 7). Locations OR and SP are situated in heavily urbanised areas on isolated peninsulas within the lake, so they were also identified as areas of conservation concern.

Subset of thin biological corridors identified with least cost paths (black lines) that require conservation for permitting the continued gene flow of squirrel gliders. Above: Fishery Point Road (Bonnells Bay) leading to squirrel glider location SP. Below: Fernleigh Track (Whitebridge) and vegetation west of Fernleigh Track (Charlestown) connecting squirrel glider locations FERNS and OCN (south) with FERNN, DUD and GSCA (north). Yellow dots indicate trapping locations where squirrel glider samples were collected
Our study not only identified corridors, but it also investigated possible barriers to dispersal. A 30-meter-wide powerline easement and the corresponding fragmentation of habitat did not seem to impact squirrel glider movement in location WR. Empirical movement data from GPS collars also confirmed that squirrel gliders can cross two lane roads in our study area (Meyer et al. 2021). However, Holt and Brady (2011) have reported the reluctance of female Mahogany gliders (Petaurus gracilis) to cross 30-meter powerline easements, so further research is needed to confirm that the crossing observed by a female squirrel glider in this study is not an anomaly. Additionally, the power poles at WR were wooden and attention should be given to areas that use metal power poles, as the squirrel gliders in this study may have been using the wooden poles as steppingstones across the landscape. It is not only powerlines that create gaps between habitat patches; roads create obstacles as well. Sections of the Pacific Highway in the northeast area of Lake Macquarie LGA have mature trees in the median strip which is extremely valuable for the gene flow of squirrel glider populations on either side (e.g. the town Bennetts Green). These trees should be conserved since Van der Ree et al. (2010) radio tracked squirrel gliders and found 67% of them crossed highways when trees were in the median strip, while only 6% (one male) crossed a highway when there were no trees (Van der Ree et al. 2010). As we did not capture squirrel gliders in any of the trapping locations west of the Pacific Motorway, the genetic effects of the Pacific Motorway on squirrel glider populations remain unclear. This said, sugar glider populations experienced genetic differentiation because of the Motorway (Knipler et al., unpublished data), and since squirrel gliders have experienced higher levels of genetic structure in the Lake Macquarie LGA then they may also be experiencing genetic differentiation on either side of this road, especially because there are no mature trees in the median strip. Structures such as glider poles (Ball and Goldingay 2008; Goldingay et al. 2019) should therefore be installed to assist the gene flow of sugar gliders, which would also help the squirrel gliders that inhabit the same area.
With our filtered SNP data, we identified corridors that could potentially enhance gene flow of squirrel gliders. Landscape genomics is a rapidly growing field that presents tremendous potential to estimate functional connectivity (Balkenhol et al. 2017). Genetically derived connectivity estimates reflect past landscape permeability due to the time it takes to detect barriers on species gene flow (Cushman et al. 2013), and thus does not necessarily detect current gene flow in a rapidly evolving landscape such as Lake Macquarie LGA. Furthermore, genetic data do not directly convey how animals move through the landscape. Hence, for conservation purposes, it is often recommended to complement connectivity analyses with other sets of empirical data. Movement data in particular would enable us to examine how animals respond to landscape features as they disperse (Cushman 2013), and would provide a more complete picture of the resources needed by squirrel gliders as they cope with habitat change.
We here used LCP to estimate functional connectivity, which assumes animals have perfect knowledge of the landscape, and therefore move across the landscape in an optimal way. In contrast, circuit-theory connectivity is based on random walk and applies concepts related to flow of charge through an electrical circuit to the movement of individuals through the landscape. The output of such models is the probability of movement across each pixel of the landscape and incorporates all possible pathways across the area studied (McRae et al. 2008). As squirrel gliders are unlikely to have a complete knowledge of their surroundings when dispersing, future studies using circuit-theory modelling may be useful to confirm our findings.
Continued fragmentation is the greatest risk to squirrel gliders in our study area, particularly as the effects of urbanisation are exacerbated by the biogeographical barrier of the Lake Macquarie. Conservation of remaining forest habitat should be a priority for the long-term survival of squirrel gliders in this area. Moreover, thin remnant corridors need to be conserved to promote gene flow and connectivity of the species populations in the urban matrix. If unsuccessful, squirrel glider populations may experience further isolation which could in turn lead to a loss of genetic diversity through genetic drift. The Lake Macquarie LGA is a stronghold for the threatened species in Australia, and as such this location warrants priority conservation management. High levels of genetic structure and population differentiation also warrants proactive conservation management in response. Finally, considering the difficulty in collecting large sample sizes of threatened species, the use of genome-wide SNPs as we have undertaken, has allowed a thorough investigation into the fine-scale structure of squirrel gliders in the Lake Macquarie LGA. This data is vital and will prove valuable for conservation management decisions in the local area.
Availability of data and material
DArTseq data and mtDNA sequences are available from the authors upon request.
Code Availability
Not applicable.
References
Adriaensen F, Chardon JP, De Blust G, Swinnen E, Villalba S, Gulinck H, Matthysen E (2003) The application of ‘least-cost’ modelling as a functional landscape model. Landsc Urban Plann 64:233–247. doi:https://doi.org/10.1016/S0169-2046(02)00242-6
Arif IA, Khan HA, Bahkali AH, Homaidan A, Farhan AAA, Sadoon AHA, Shobrak M (2011) DNA marker technology for wildlife conservation. Saudi J Biol Sci 18:219–225. doi:https://doi.org/10.1016/j.sjbs.2011.03.002
Avise JC (2000) ‘Phylogeography: the history and formation of species’. Harvard University Press, Cambridge.)
Baden AL, Holmes SM, Johnson SE, Engberg SE, Louis EE, Bradley BJ (2014) Species-level view of population structure and gene flow for a critically endangered primate (Varecia variegata). Ecol Evol 4:2675–2692. doi:https://doi.org/10.1002/ece3.1119
Balkenhol N, Cushman SA, Storfer AT, Waits LP (2015) Introduction to Landscape Genetics - Concepts, Methods, Applications. Landscape Genetics: Concepts, Methods, Applications, 1–8. doi:https://doi.org/10.1002/9781118525258.ch01
Balkenhol N, Dudaniec RY, Krutovsky KV, Johnson JS, Cairns DM, Segelbacher G, Selkoe KA, von der Heyden S, Wang IJ, Selmoni O, Joost S (2017) Landscape Genomics: Understanding Relationships Between Environmental Heterogeneity and Genomic Characteristics of Populations. In pp. 261–322. (Springer, Cham.) doi:https://doi.org/10.1007/13836_2017_2
Ball T, Adams E, Goldingay RL (2009) Diet of the squirrel glider in a fragmented landscape near Mackay, central Queensland. Australian J Zool 57:295–304. doi:https://doi.org/10.1071/ZO08095
Ball TM, Goldingay RL (2008) Can wooden poles be used to reconnect habitat for a gliding mammal? Landsc Urban Plann 87:140–146. doi:https://doi.org/10.1016/j.landurbplan.2008.05.007
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. doi:https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barratt EM, Gurnell J, Malarky G, Deaville R, Bruford MW (1999) Genetic structure of fragmented populations of red squirrel (Sciurus vulgaris) in the UK. Mol Ecol 8:55–63. doi:https://doi.org/10.1046/j.1365-294X.1999.00771.x
Bell S, Driscoll C, Survey EF (2016) Volume 1: Vegetation Mapping Report, Lake Macquarie Local Government Area Stages 1–6
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N, Mendes MM, Müller FK, Ogilvie NF, Plessis HA, du Popinga L, Rambaut A, Rasmussen A, Siveroni D, Suchard I, Wu MA, Xie C-H, Zhang D, Stadler C, Drummond AJ (2019) Analysis., BEAST 2.5: An advanced software platform for Bayesian evolutionary. PLoS Comput Biol 15:e1006650
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinf Appl 23:2633–2635. doi:https://doi.org/10.1093/bioinformatics/btm308
Bradshaw CJA (2012) Little left to lose: Deforestation and forest degradation in Australia since European colonization. J Plant Ecol 5:109–120. doi:https://doi.org/10.1093/jpe/rtr038
Brearley G, McAlpine C, Bell S, Bradley A (2011) Squirrel glider home ranges near urban edges in eastern Australia. J Zool 285:256–265. doi:https://doi.org/10.1111/j.1469-7998.2011.00837.x
Brumfield RT, Beerli P, Nickerson DA, Edwards SV (2003) The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol Evol 18:249–256. doi:https://doi.org/10.1016/S0169-5347(03)00018-1
Cullingham CI, Miller JM, Peery RM, Dupuis JR, Malenfant RM, Gorrell JC, Janes JK (2020) Confidently identifying the correct K value using the ∆K method: When does K = 2? Mol Ecol 29:862–869. doi:https://doi.org/10.1111/mec.15374
Cushman SA (2013) Biological corridors and connectivity. ‘Key Topics in Conservation Biology 2’. Wiley-Blackwell, Hoboken, NJ.), pp 384–404
Cushman SA, Lewis JS (2010) Movement behavior explains genetic differentiation in American black bears. Landscape Ecol 25:1613–1625. doi:https://doi.org/10.1007/s10980-010-9534-6
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214. doi:https://doi.org/10.1111/1755-0998.12157
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol Biol Evol 22:1185–1192. doi:https://doi.org/10.1093/MOLBEV/MSI103
Dudaniec RY, Wilmer W, Hanson J, Warren JO, Bell M, Rhodes JR (2016) Dealing with uncertainty in landscape genetic resistance models: A case of three co-occurring marsupials. Mol Ecol 25:470–486. doi:https://doi.org/10.1111/mec.13482
Dussex N, Taylor HR, Stovall WR, Rutherford K, Dodds KG, Clarke SM, Gemmell NJ (2018) Reduced representation sequencing detects only subtle regional structure in a heavily exploited and rapidly recolonizing marine mammal species. Ecol Evol 8:8736–8749. doi:https://doi.org/10.1002/ece3.4411
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. doi:https://doi.org/10.1007/s12686-011-9548-7
Eco Logical Australia Pty Ltd (2003) Lower Hunter & Central Coast Regional Biodiversity Conservation Strategy Technical Report 2003 Digital Aerial Photo Interpretation & Updated Extant Vegetation Community Map
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. doi:https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fallding M (2015) Lake Macquarie Squirrel Glider Planning and Managing Guidelines 2015. (Lake Macquarie City Council, Lake Macquarie)
Francis MJ, Spooner PG, Matthews A (2015) The influence of urban encroachment on squirrel gliders (Petaurus norfolcensis): Effects of road density, light and noise pollution. Wildl Res 42:324–333. doi:https://doi.org/10.1071/WR14182
Frankham R, Ballou JD, Briscoe DA (2002) ‘Introduction to Conservation Genetics’. (Cambridge University Press: Cambridge.) doi:https://doi.org/10.1016/j.foreco.2003.12.001
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925. doi:https://doi.org/10.1093/GENETICS/147.2.915
Goldingay RL, Harrisson KA, Taylor AC, Ball TM, Sharpe DJ, Taylor BD (2013) Fine-scale genetic response to landscape change in a gliding mammal. PLoS ONE 8:1–10. doi:https://doi.org/10.1371/journal.pone.0080383
Goldingay RL, Sharpe DJ (2004) How do we conserve the squirrel glider in Brisbane’s urban matrix? In ‘Conservation of Australia’s Forest Fauna ’. pp. 663–677 Available at: http://publications.rzsnsw.org.au/doi/pdf/https://doi.org/10.7882/FS.2004.038
Goldingay RL, Sharpe DJ, Dobson MDJ (2010) Variation in the home-range size of the squirrel glider (Petaurus norfolcensis). Australian Mammalogy 32:183–188. doi:https://doi.org/10.1071/AM10006
Goldingay RL, Taylor BD (2009) Gliding performance and its relevance to gap crossing by the squirrel glider (Petaurus norfolcensis). Australian J Zool 57:99–104. doi:https://doi.org/10.1071/ZO09003
Goldingay RL, Taylor BD, Parkyn JL (2019) Use of tall wooden poles by four species of gliding mammal provides further proof of concept for habitat restoration. Australian Mammalogy 41:255. doi:https://doi.org/10.1071/AM18008
Gracanin A, Gracanin V, Mikac KM (2019) The selfie trap: A novel camera trap design for accurate small mammal identification. Ecol Manag Restor 20:156–158. doi:https://doi.org/10.1111/emr.12345
Grant WS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. In ‘Journal of Heredity’. pp. 415–426 doi:https://doi.org/10.1093/jhered/89.5.415
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour 18:691–699. doi:https://doi.org/10.1111/1755-0998.12745
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41, 95–98. doi:citeulike-article-id:691774
Holt S, Brady M, for Ingham-Tully Transmission Line Upgrade DOCUMENT CONTROL SUMMARY REPORT AND CLIENT DETAILS Title (2011) Mahogany Glider Management Plan : Mahogany Glider Management Plan for Ingham-Tully Transmission Line Upgrade-Addendum Report Client: Powerlink Queensland. Available at: www.biotropica.com.au [accessed 16 May 2021]
Irwin DM, Kocher TD, Wilson AC (1991) Evolution of the cytochrome b gene of mammals. J Mol Evol 32:128–144. doi:https://doi.org/10.1007/BF02515385
Jombart T (2008) Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi:https://doi.org/10.1093/bioinformatics/btn129
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity arrays technology: A generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89. doi:https://doi.org/10.1007/978-1-61779-870-2_5
Kjeldsen SR, Raadsma HW, Leigh KA, Tobey JR, Phalen D, Krockenberger A, Ellis WA, Hynes E, Higgins DP, Zenger KR (2019) Genomic comparisons reveal biogeographic and anthropogenic impacts in the koala (Phascolarctos cinereus): a dietary-specialist species distributed across heterogeneous environments. Heredity 122:525–544. doi:https://doi.org/10.1038/s41437-018-0144-4
Kjeldsen SR, Zenger KR, Leigh K, Ellis W, Tobey J, Phalen D, Melzer A, FitzGibbon S, Raadsma HW (2016) Genome-wide SNP loci reveal novel insights into koala (Phascolarctos cinereus) population variability across its range. Conserv Genet 17:337–353. doi:https://doi.org/10.1007/s10592-015-0784-3
Knipler ML, Dowton M, Mikac KM (2021) Genome-Wide SNPs Detect Hybridisation of Marsupial Gliders (Petaurus breviceps breviceps × Petaurus norfolcensis) in the Wild. Genes 12:1327. doi: https://doi.org/10.3390/genes12091327
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC, Pääbo S, Villablanca FX, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86:6196–6200. doi:https://doi.org/10.1073/pnas.86.16.6196
Kumar S, Banks TW, Cloutier S (2012) SNP Discovery through Next-Generation Sequencing and Its Applications. International journal of plant genomics 2012, 831460. doi:https://doi.org/10.1155/2012/831460
Lah L, Trense D, Benke H, Gunnlaugsson Þ, Lockyer C, Rk AO, ¨ Z, Rk BO, ¨ Z, Pawliczka I, Roos A, Siebert U, Víkingsson K, Tiedemann R (2016) Spatially Explicit Analysis of Genome-Wide SNPs Detects Subtle Population Structure in a Mobile Marine Mammal, the Harbor Porpoise. doi:https://doi.org/10.1371/journal.pone.0162792
Lampila S, Kvist L, Wistbacka R, Orell M (2009) Genetic diversity and population differentiation in the endangered Siberian flying squirrel (Pteromys volans) in a fragmented landscape. Eur J Wildl Res 55:397–406. doi:https://doi.org/10.1007/s10344-009-0259-2
Lancaster ML, Cooper SJB, Carthew SM (2016) Genetic consequences of forest fragmentation by agricultural land in an arboreal marsupial. Landscape Ecol 31:655–667. doi:https://doi.org/10.1007/s10980-015-0271-8
Larsen PA, Marchán-Rivadeneira MR, Baker RJ (2013) Speciation Dynamics of the Fruit-Eating Bats (Genus Artibeus): With Evidence of Ecological Divergence in Central American Populations. ‘Bat Evolution, Ecology, and Conservation’. Springer New York, New York, NY, pp 315–339. doi:https://doi.org/10.1007/978-1-4614-7397-8_16
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:https://doi.org/10.1093/bioinformatics/btp187
Lindenmayer D (2002) ‘Gliders of Australia: a natural history’. (UNSW Press.) Available at: https://openresearch-repository.anu.edu.au/handle/1885/92545 [accessed 16 August 2017]
Liu N, Chen L, Wang S, Oh C, Zhao H (2005) Comparison of single-nucleotide polymorphisms and microsatellites in inference of population structure. BMC Genet 6. doi:https://doi.org/10.1186/1471-2156-6-S1-S26
Malekian M, Cooper SJB, Norman JA, Christidis L, Carthew SM (2010) Molecular systematics and evolutionary origins of the genus Petaurus (Marsupialia: Petauridae) in Australia and New Guinea. Mol Phylogenet Evol 54:122–135. doi:https://doi.org/10.1016/J.YMPEV.2009.07.026
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: Combining landscape ecology and population genetics. Trends in Ecology and Evolution 18:189–197. doi:https://doi.org/10.1016/S0169-5347(03)00008-9
Mawbey R (1989) A New Trap Design for the Capture of Sugar Gliders, Petaurus-Breviceps. Wildl Res 16:425–428
McRae BH, Dickson BG, Keitt TH, Shah VB (2008) Using circuit theory to model connectivity in ecology, evolution, and conservation. Ecology 89:2712–2724. doi:https://doi.org/10.1890/07-1861.1
Meredith RW, Mendoza MA, Roberts KK, Westerman M, Springer MS (2010) A Phylogeny and Timescale for the Evolution of Pseudocheiridae (Marsupialia: Diprotodontia) in Australia and New Guinea. Journal of Mammalian Evolution 2010 17:2 17, 75–99. doi:https://doi.org/10.1007/S10914-010-9129-7
Meyer NFV, King JP, Mahony M, Clulow J, Beranek C, Reedman C, Balkenhol N, Hayward MW (2021) Large area used by squirrel gliders in an urban area, uncovered using GPS telemetry. Ecol Evol 11:7147–7153. doi:https://doi.org/10.1002/ECE3.7644
Milanesi P, Holderegger R, Caniglia R, Fabbri E, Randi E (2016) Different habitat suitability models yield different least-cost path distances for landscape genetic analysis. Basic Appl Ecol 17:61–71. doi:https://doi.org/10.1016/j.baae.2015.08.008
Niche Environment and Heritage (2013) Distribution, habitat and conservation of the Squirrel Glider near Forster, NSW. OEH, Parramatta Office
NSW Government (2007) NSW Landuse 2007 - Data.NSW. Available at: https://data.nsw.gov.au/data/dataset/nsw-landuseac11c/resource/e641e722-96c5-4519-874a-e148a496fac6?inner_span=True [accessed 6 May 2021]
NSW Office of Environment and Heritage (2021) BioNet Atlas. Available at: https://www.environment.nsw.gov.au/atlaspublicapp/ui_modules/atlas_/atlassearch.aspx
Pavlova A, Walker FM, Van der Ree R, Cesarini S, Taylor AC (2010) Threatened populations of the Australian squirrel glider (Petaurus norfolcensis) show evidence of evolutionary distinctiveness on a Late Pleistocene timescale. Conserv Genet 11:2393–2407. doi:https://doi.org/10.1007/s10592-010-0125-5
Payne C (2016) The effects of habitat fragmentation and vegetation type on forest structure and arboreal marsupial populations, with particular focus on the squirrel glider (Petaurus norfolcensis), in an urban landscape. Honours thesis, University of Wollongong, Wollongong NSW
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard J, Wen X, Falush D (2003) Documentation for STRUCTURE software: version 2.3 | Request PDF. Available at: https://www.researchgate.net/publication/252278965_Documentation_for_STRUCTURE_software_version_23 [accessed 12 March 2021]
Quin DG (1995) Population ecology of the squirrel glider (Petaurus norfolcensis) and the sugar glider (P. breviceps) (Maruspialia: Petauridae) at Limeburners Creek, on the central north coast of New South Wales. Wildl Res 22:471–505. doi:https://doi.org/10.1071/WR9950471
R Core Team (2015) R: a language and environment for statistical computing. Available at: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing [accessed 1 April 2021]
Rambaut A (2019) Figtree v1.4.4. Available at: http://tree.bio.ed.ac.uk/software/figtree/ [accessed 13 November 2021]
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst Biol 67:901–904. doi:https://doi.org/10.1093/SYSBIO/SYY032
Van der Ree R (2002) The population ecology of the squirrel glider (Petaurus norfolcensis) within a network of remnant linear habitats. Wildl Res 29:329–340. doi:https://doi.org/10.1071/WR01095
Van der Ree R, Bennett AF (2003) Home range of the squirrel glider (Petaurus norfolcensis) in a network of renant linear habitats. J Zoological Soc Lond 259:327–336. doi:https://doi.org/10.1017/s0952836902003229
Van der Ree R, Bennett AF, Gilmore DC (2004) Gap-crossing by gliding marsupials: Thresholds for use of isolated woodland patches in an agricultural landscape. Biol Conserv 115:241–249. doi:https://doi.org/10.1016/S0006-3207(03)00142-3
Van der Ree R, Cesarini S, Sunnucks P, Moore JL, Taylor A (2010) Large gaps in canopy reduce road crossing by a gliding mammal. Ecol Soc 15:35
Schneider S, Excoffier L (1999) Estimation of Past Demographic Parameters From the Distribution of Pairwise Differences When the Mutation Rates Vary Among Sites: Application to Human Mitochondrial DNA. Genetics 152:1079–1089
Schultz AJ, Cristescu RH, Littleford-Colquhoun BL, Jaccoud D, Frère CH (2018) Fresh is best: Accurate SNP genotyping from koala scats. Ecol Evol 8:3139–3151. doi:https://doi.org/10.1002/ece3.3765
Sgarlata GM, Salmona J, Razanaparany TP, Joseph C, Rabarivola FJ, Rasolondraibe E, Andriaholinirina N, Lafosse S, Chikhi L, Manni F, Bayart F (2016) Mitochondrial Genetic Diversity in the Crowned Sifaka (Propithecus coronatus) in a Fragmented Landscape. Primate Conserv 30:39–57
Sharma S, Dutta T, Maldonado JE, Wood TC, Panwar HS, Seidensticker J (2013) Forest corridors maintain historical gene flow in a tiger metapopulation in the highlands of central India. Proceedings of the Royal Society B: Biological Sciences 280. doi:https://doi.org/10.1098/rspb.2013.1506
Sharpe D (2004) ‘Effect of flower failure on a population of squirrel gliders (Petaurus norfolcensis) in northeast New South Wales. In “The Biology of Australian Possums and Gliders”. (Eds RL Goldingay & SM Jackson)’. (Surrey Beatty: Sydney.)
Sharpe D, Goldingay RL (2007) Home range of the Australian squirrel glider, Petaurus norfolcensis (Diprotontia). J Mammal 88:1515–1522. doi:https://doi.org/10.1644/06-MAMM-A-404R1.1
Shi W, Ayub Q, Vermeulen M, Shao RG, Zuniga S, Van Der Gaag K, De Knijff P, Kayser M, Xue Y, Tyler-Smith C (2010) A worldwide survey of human male demographic history based on Y-SNP and Y-STR data from the HGDP-CEPH populations. Mol Biol Evol 27:385–393. doi:https://doi.org/10.1093/molbev/msp243
Smith A (2002) Squirrel Glider (Petaurus norfolcensis) Conservation Management Plan. Wyong Shire Prepared by Andrew Smith for Wyong Shire Council
Smith AP, Murray M (2003) Habitat requirements of the squirrel glider (Petaurus norfolcensis) and associated possums and gliders on the New South Wales central coast. Wildl Res 30:291–301. doi:https://doi.org/10.1071/WR01115
Soanes K, Vesk PA, Van der Ree R (2015) Monitoring the use of road-crossing structures by arboreal marsupials: Insights gained from motion-triggered cameras and passive integrated transponder (PIT) tags. Wildl Res 42:241–256. doi:https://doi.org/10.1071/WR14067
Subramanian S (2016) The effects of sample size on population genomic analyses – implications for the tests of neutrality. BMC Genomics 17. doi:https://doi.org/10.1186/S12864-016-2441-8
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. doi:https://doi.org/10.1093/GENETICS/123.3.585
Taylor AC, Walker FM, Goldingay RL, Ball T, van der Ree R (2011) Degree of landscape fragmentation influences genetic isolation among populations of a gliding mammal. PLoS ONE 6. doi:https://doi.org/10.1371/journal.pone.0026651
White LC, Moseby KE, Thomson VA, Donnellan SC, Austin JJ (2018) Long-term genetic consequences of mammal reintroductions into an Australian conservation reserve. Biol Conserv 219:1–11. doi:https://doi.org/10.1016/j.biocon.2017.12.038
Whitlock MC, Lotterhos KE (2015) Reliable Detection of Loci Responsible for Local Adaptation: Inference of a Null Model through Trimming the Distribution of F(ST). Am Nat 186(Suppl 1):S24–S36. doi:https://doi.org/10.1086/682949
Winning G, King J (2008) A new trap design for capturing squirrel gliders and sugar gliders. Australian Mammology 29:245–249
Woinarski J, Burbidge AA, Harrison P, Commonwealth Scientific and Industrial Research Organization (Australia) (2014) ‘The action plan for Australian mammals 2012’. CSIRO publishing, Collingwood, VIC.)
Wright B, Farquharson KA, McLennan EA, Belov K, Hogg CJ, Grueber CE (2019) From reference genomes to population genomics: Comparing three reference-aligned reduced-representation sequencing pipelines in two wildlife species. BMC Genomics 20:453. doi:https://doi.org/10.1186/s12864-019-5806-y
Zachos FE, Frantz AC, Kuehn R, Bertouille S, Colyn M, Niedziałkowska M, Pérez-González J, Skog A, Sprěm N, Flamand MC (2016) Genetic Structure and Effective Population Sizes in European Red Deer (Cervus elaphus) at a Continental Scale: Insights from Microsatellite DNA. J Hered 107:318–326. doi:https://doi.org/10.1093/JHERED/ESW011
Zeller KA, McGarigal K, Whiteley AR (2012) Estimating landscape resistance to movement: A review. Landscape Ecol 27:777–797. doi:https://doi.org/10.1007/s10980-012-9737-0
Zhian Kamvar MN (2021) Package ‘poppr’ Type Package Title Genetic Analysis of Populations with Mixed Reproduction Encoding UTF-8 Depends R ( > = 2.15.1), adegenet ( > = 2.0.0). https://doi.org/10.7717/peerj.281
Zimmerman SJ, Aldridge CL, Oyler-Mccance SJ (2020) An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genomics 21. doi:https://doi.org/10.1186/S12864-020-06783-9
Acknowledgements
We acknowledge the Awabakal people, the Traditional Custodians of the land where our research took place. Funding was provided by a Lake Macquarie City Council Environmental Research grant (KMM and JC), University of Wollongong Centre for Sustainable Ecosystem Solutions advancement grant (MLK) and Paddy Pallin Research Grant from the Royal Zoological Society of New South Wales (NM). The authors thank Lake Macquarie City Council and NSW National Parks & Wildlife Service for providing access to land on which gliders were trapped. Additionally, we thank students who assisted in collecting samples, particularly Reannan Honey, John-Paul King, Sarah Downey and Kate Maslen.
Funding
Lake Macquarie City Council, Environmental Grants (2018–2020); UOW Centre for Sustainable Ecosystem Solutions, research student support grant (2020), Paddy Pallin Research Grant from the Royal Zoological Society of New South Wales.
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
Monica Knipler, Mark Dowton, John Clulow and Katarina Mikac contributed to the study conception and design. Material preparation and sample collection were performed by Monica Knipler and Ninon Meyer, and analyses were performed by Monica Knipler with advice from Mark Dowton and Katarina Mikac. The first draft of the manuscript was written by Monica Knipler and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest/Competing interests
The authors declare no competing interest.
Animal research
Squirrel glider genetic samples were collected under University of Wollongong Animal Ethics permits AE15/11 and AE19/02 and NSW Scientific License SL101587 and SL101968, the University of Newcastle Animal Care and Ethics Committee (A-2015-510), and NSW Scientific License SL100190.
Consent to participate
Not applicable.
Consent for publication
All authors consent to the submission of this article to Conservation Genetics.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Knipler, M.L., Dowton, M., Clulow, J. et al. Genome-wide SNPs detect fine-scale genetic structure in threatened populations of squirrel glider Petaurus norfolcensis. Conserv Genet 23, 541–558 (2022). https://doi.org/10.1007/s10592-022-01435-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01435-9