
Abstract
The increasing prevalence of neurological disorders such as Alzheimer’s, Parkinson’s, and multiple sclerosis presents a significant global health challenge. Despite extensive research, the precise mechanisms underlying these conditions remain elusive, with current treatments primarily addressing symptoms rather than root causes. Emerging evidence suggests that gut permeability and the kynurenine pathway are involved in the pathogenesis of these neurological conditions, offering promising targets for novel therapeutic and preventive strategies. Gut permeability refers to the intestinal lining’s ability to selectively allow essential nutrients into the bloodstream while blocking harmful substances. Various factors, including poor diet, stress, infections, and genetic predispositions, can compromise gut integrity, leading to increased permeability. This condition facilitates the translocation of toxins and bacteria into systemic circulation, triggering widespread inflammation that impacts neurological health via the gut–brain axis. The gut–brain axis (GBA) is a complex communication network between the gut and the central nervous system. Dysbiosis, an imbalance in the gut microbiota, can increase gut permeability and systemic inflammation, exacerbating neuroinflammation—a key factor in neurological disorders. The kynurenine pathway, the primary route for tryptophan metabolism, is significantly implicated in this process. Dysregulation of the kynurenine pathway in the context of inflammation leads to the production of neurotoxic metabolites, such as quinolinic acid, which contribute to neuronal damage and the progression of neurological disorders. This narrative review highlights the potential and progress in understanding these mechanisms. Interventions targeting the kynurenine pathway and maintaining a balanced gut microbiota through diet, probiotics, and lifestyle modifications show promise in reducing neuroinflammation and supporting brain health. In addition, pharmacological approaches aimed at modulating the kynurenine pathway directly, such as inhibitors of indoleamine 2,3-dioxygenase, offer potential avenues for new treatments. Understanding and targeting these interconnected pathways are crucial for developing effective strategies to prevent and manage neurological disorders.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rising prevalence of neurological disorders (NDs) such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS) poses a significant challenge to public health globally (Brown 2019). Despite extensive research, the exact mechanisms underlying these disorders remain elusive, with current treatments primarily focused on symptom management rather than addressing the root causes. Increasing evidence suggests that gut permeability, often referred to as “leaky gut”, and the kynurenine pathway (KP) plays crucial roles in the pathogenesis of these neurological conditions, presenting potential targets for novel preventive and therapeutic strategies (Erickson et al. 2012; Konsman 2022).
Gut permeability refers to the ability of the intestinal lining to prevent harmful substances from entering the bloodstream while allowing essential nutrients to pass through. A healthy gut barrier is critical for maintaining overall health, including neurological health (Allam-Ndoul et al. 2020). However, various factors such as poor diet, stress, infections, and genetic predispositions can compromise the integrity of the gut lining, leading to increased permeability. This condition allows toxins, bacteria, and other inflammatory substances to translocate from the gut into the systemic circulation, triggering widespread inflammation (Brown 2019).
The relationship between gut permeability and neurological disorders is mediated through the gut–brain axis (GBA), a complex communication network linking the gut and the central nervous system (CNS) (O’Mahony et al. 2015). This bidirectional pathway involves neural, hormonal, and immune signalling mechanisms, allowing the gut microbiota to influence brain function and vice versa. Dysbiosis, or an imbalance in the gut microbiota, can lead to increased gut permeability and systemic inflammation, which in turn can exacerbate neuroinflammation—a key factor in the development and progression of NDs (Dinan & Cryan 2017a, b).
One of the critical pathways implicated in this process is the KP, the primary route for tryptophan metabolism in the body (Mor et al. 2021). Under normal conditions, this pathway helps regulate immune responses and neurotransmitter production. However, in the context of increased gut permeability and systemic inflammation, the activation of the KP can become dysregulated. This dysregulation leads to the production of neurotoxic metabolites such as quinolinic acid, which can induce oxidative stress and excitotoxicity in the CNS, contributing to neuronal damage and the progression of neurological disorders (Hestad et al. 2022).
The KP’s involvement in neuroinflammation is particularly significant because it represents a potential point of intervention. By modulating this pathway, it may be possible to reduce the production of harmful metabolites and mitigate their effects on the brain. For instance, targeting the enzyme indoleamine 2,3-dioxygenase (IDO), which catalyses the initial step in the KP, could help reduce the levels of neurotoxic compounds and support neuronal health (Kennedy et al. 2017).
Moreover, the gut microbiota itself plays a crucial role in modulating the KP. Certain beneficial bacteria can influence the production of KP metabolites, thereby affecting neuroinflammatory processes. Probiotics and prebiotics, which help maintain a healthy gut microbiota, have shown promise in reducing gut permeability and systemic inflammation, potentially offering a protective effect against neurological disorders (Dinan et al. 2013; Sarkar et al. 2016; Xiang et al. 2022). These findings highlight the importance of maintaining a balanced gut microbiota through diet, lifestyle, and targeted interventions as a strategy for preventing and managing NDs (Miller et al., 2020).
In addition to dietary and microbial interventions, other preventive strategies may include pharmacological approaches aimed at modulating the KP directly. Research into specific inhibitors of enzymes involved in this pathway, such as IDO inhibitors, is ongoing and holds potential for developing new treatments for NDs. These inhibitors could help restore balance in the KP, reducing the burden of neurotoxic metabolites and supporting overall brain health (Platten et al. 2019).
Microbiota and Gut Permeability
The concept of ‘microbiota’ originated in the early 1900s, recognising that a myriad of microorganisms, including bacteria, yeasts, and viruses, inhabit various human body sites such as the gut, skin, lungs, and oral cavity (Ursell et al. 2012). The precise timing of microbiota initiation, whether before or after birth, remains debated. Nevertheless, it is widely accepted that postnatal factors substantially influence the microbiome’s development (Matsuda et al. 2004). The GI tract, with its diverse microbial habitats, hosts the densest microbial population, comprising over 1500 bacterial species across more than 50 phyla (Dinan & Cryan 2017b). The predominant phyla include Bacteroidetes and Firmicutes, followed by Proteobacteria, Fusobacteria, Tenericutes, Actinobacteria, and Verrucomicrobia, accounting for approximately 90% of the microbial population (Louwies et al. 2020).
The intricate relationship between the immune system and the gut microbiota has evolved into a finely tuned mutualistic interaction essential for maintaining homeostasis. This balance ensures effective host immunity, preventing commensal microbes from exploiting resources excessively while maintaining tolerance to benign stimuli. However, various disruptions, such as antibiotic usage, dietary changes, and environmental pollutants, can destabilise this balance, leading to significant health implications. These disruptions can impair the interfaces between the host and microbiome, altering immune responses and potentially leading to systemic spread of commensal microbes, increased susceptibility to pathogenic invasion, and inappropriate immune reactions (Allam-Ndoul et al. 2020; Brown 2019; Morris et al. 2015).
The gastrointestinal (GI) tract, a complex organ system responsible for nutrient digestion and absorption, also plays a critical role in maintaining a barrier that protects the body from harmful substances. The gut lining, or intestinal epithelium, is central to this function. It serves as a selectively permeable membrane, allowing essential nutrients to pass while preventing harmful pathogens and toxins from entering the body (Takiishi et al. 2017). The intestinal epithelial cells (IECs), primarily enterocytes, form the gut lining and regulate the trans-epithelial movement of substances. This lining is reinforced by a complex array of junction proteins, including tight junctions, adherens junctions, gap junctions, and desmosomes, which ensure the structural integrity and function of the gut barrier (Fig. 1) (Schoultz & Keita 2020; Wells et al. 2017). Tight junctions are key regulators of the paracellular pathway, controlling the flow of ions, water, and various macromolecules between epithelial cells (Suzuki 2020).

The molecular composition of the tight junction (TJ). TJs are constituted by the transmembrane proteins occludin, claudins, and junctional adhesion molecule 1 (JAM-1), which seal the paracellular space and connect TJ to the actin cytoskeleton via interaction with proteins from the zona occludens (ZO) family. (With kind permission from Springer Science + Business Media: Histochem. Cell Biol., Tight junctions and the modulation of barrier function in disease, 130(1), 2008, 55, Förster, C., Copyright 2008)
The gut microbiota employs various defence mechanisms to prevent pathogen overgrowth and resultant damage or infection. One such mechanism is colonisation resistance, where both commensal and pathogenic microorganisms compete for resources and functional space, often mediated by quorum sensing (Takiishi et al. 2017). IECs not only form a physical barrier but also engage in active immune responses. The mucus layer produced by goblet cells adds an additional protective layer, preventing direct contact between luminal bacteria and the epithelial surface (Tlaskalová-Hogenová et al. 2011).
The epithelial barrier is a multilayer system that provides both physical and functional protection. Key components include luminal intestinal alkaline phosphatase (IAP), which dephosphorylates bacterial endotoxin lipopolysaccharide (LPS) to detoxify it; the mucus layer, which acts as a physical barrier preventing interactions between gut bacteria and IECs; tight junctions, which limit the paracellular transport of bacteria and their products to systemic circulation; and antibacterial proteins and immunoglobulin A (IgA), secreted by Paneth cells and immune cells in the lamina propria, which contribute to mucosal defence(Iacob & Iacob 2019; Odenwald & Turner 2017).
The intestinal epithelium serves as an essential barrier, orchestrating selective permeability that balances nutrient absorption with the exclusion of harmful substances. This barrier function is meticulously regulated through transcellular and paracellular pathways (Fig. 1).
The transcellular pathway involves the active transport of substances directly through epithelial cells, relying on various transporters and channels embedded in cell membranes. This route is highly specific and energy-dependent, facilitating the transport of nutrients, ions, and macromolecules (Madara 1998; Günzel & Yu 2013) (Fig. 2).

a General transport pathways: Paracellular, transcellular, and transcytosis. b Transcellular membrane transport: Transport across the apical cell membrane can be via (1) passive transport, which can be via (I) simple diffusion and (II) facilitated diffusion. Facilitated diffusion can, in turn, be either channel-mediated facilitated diffusion or carrier-mediated facilitated diffusion. (2) Active transport, which can be (I) primary active transport and (II) secondary active transport. c Endocytosis/transcytosis: Transport in vesicles that can be via (1) phagocytosis, via specialised cells of the reticuloendothelial system, e.g. neutrophils and macrophages, (2) pinocytosis: Nonspecific internalisation of extracellular fluid (ECF); any dissolved solutes that happen to be in the ECF also internalised and (3) receptor-mediated endocytosis/transcytosis, a highly selective type of endocytosis/transcytosis, by which cells take up specific ligands (Laurent et al. 2016)
The paracellular pathway, on the other hand, allows passive movement of ions and small molecules between epithelial cells, regulated by tight junctions (Buckley & Turner 2018). This pathway is crucial for maintaining ionic balance and water flux in the intestinal lumen (Spadoni et al. 2015). Tight junctions are multi-protein complexes located at the apical region of epithelial cells, comprising transmembrane proteins such as claudins, occludins, and junctional adhesion molecules (JAMs), which interact with intracellular scaffold proteins like zonula occludens (ZO-1, ZO-2, ZO-3), linking tight junctions to the actin cytoskeleton (Odenwald & Turner 2017; Okumura & Takeda 2017).
The pore pathway within tight junctions allows the passage of small ions and uncharged molecules, regulated primarily by claudins forming charge-selective pores (Sturgeon & Fasano 2016). The leak pathway, which is less selective, accommodates the passage of larger molecules, including proteins and larger ions, regulated by occludin and ZO proteins (Odenwald & Turner 2017). Transepithelial resistance measures the epithelium’s ability to resist ion flow, serving as an indicator of paracellular permeability, with low TER values indicating high permeability (Schoultz & Keita 2020).
Intestinal permeability is dynamically regulated by various physiological and pathological factors. Inflammatory mediators such as pro-inflammatory cytokines (e.g. Tumour Necrosis Factor-alpha and Interferon-gamma) can disrupt tight junctions, increasing paracellular permeability. These cytokines activate signalling pathways leading to the reorganisation of tight-junction proteins and the actin cytoskeleton, resulting in increased permeability, particularly relevant in inflammatory conditions such as inflammatory bowel disease (IBD) (Nishida et al. 2018; Sturgeon & Fasano 2016).
Myosin light chain kinase (MLCK) also plays a crucial role in regulating tight-junction permeability. Activation of MLCK leads to the phosphorylation of myosin light chains, causing contraction of the actomyosin ring and increasing tight-junction permeability, a mechanism involved in conditions associated with increased intestinal permeability, such as chronic stress and infection (Buckley & Turner 2018). Dietary components and the gut microbiota significantly influence intestinal permeability. Short-chain fatty acids (SCFAs), produced by the fermentation of dietary fibres by gut bacteria, enhance barrier function by modulating the expression of tight-junction proteins, whereas high-fat diets and dysbiosis can disrupt tight-junction integrity, leading to increased permeability (Lee et al. 2018).
Oxidative stress, induced by reactive oxygen species (ROS), can damage tight-junction proteins, leading to increased permeability (Mor et al. 2021). Antioxidant mechanisms are essential for maintaining tight-junction integrity and protecting against oxidative stress-induced barrier dysfunction, particularly relevant in conditions such as IBD and metabolic disorders (Bailey et al. 2011). Hormones and neuropeptides, such as glucocorticoids and corticotropin-releasing hormone, also modulate intestinal permeability, with stress-induced release of these hormones leading to increased permeability, highlighting the intricate connection between the gut and the CNS (Zuhl et al. 2015).
The Microbiota–Gut–Brain Axis
The GBA is a bidirectional communication network orchestrated through a complex network of neurons, neurotransmitters, hormones, and immune mediators. While its significance in mental and cognitive health is widely acknowledged, the precise mechanisms through which the gut microbiota exerts influence on brain development and function remain an active area of investigation (Berding et al. 2021; Breit et al. 2018; Dinan & Cryan 2017b; Gomez-Eguilaz et al. 2019; Louwies et al. 2020). The GBA encompasses a multifaceted communication network primarily between the central and enteric nervous systems, linking the brain’s emotional and cognitive centres with intestinal function (O’Mahony et al. 2015). Both clinical and experimental research underscore that dysfunctions along the GBA could be implicated in brain and cognitive diseases (Dinan & Cryan 2017b). Alterations in the gut microbiota can modulate the peripheral and CNS, influencing brain stimulation and cognitive functioning via various signalling pathways (Dinan & Cryan 2017a).
Neuronal pathways form a critical component of the GBA. The vagus nerve, extending from the brainstem to the gut, is pivotal in these pathways (Dinan & Cryan 2017b). It detects sensory signals from the gut and conveys them to the CNS, involving the activation of mechanoreceptors and chemoreceptors responsive to chemical stimuli, including hormones, neurotransmitters, and metabolites produced by EECs (O’Mahony et al. 2015). The ENS, often referred to as the “second brain”, contains an extensive neuronal network regulating gut functions and is influenced by the gut microbiota, impacting gut motility and intestinal barrier function (Zhu et al. 2024).
Both the CNS and the gut microbiota directly affect, and are affected by, the immune system. The gut microbiota is crucial in modulating the development and function of the peripheral immune system (C. Wang et al. 2019). It is also integral to the healthy development, maturation, and activation of microglia, which are the innate immune cells in the brain (Erny et al. 2015). The activation of microglia is believed to depend on signals from microbial metabolism, as evidenced by the restoration of microglial morphology and function in germ-free mice treated with bacterial derived (Erny et al. 2015).
The gut microbiota’s interaction with the brain is also mediated through the systemic immune system via circulating cytokines (Campbell et al. 2014). Changes in systemic immunity can lead to altered immune signalling within the brain, resulting in symptoms such as loss of appetite, irritability, low mood, loss of motivation, social withdrawal, fatigue, and impaired attention (Anderson et al. 2021; Chen et al. 2021; Connell et al. 2017; Huang & Wu 2021). Cytokines and chemokines produced by brain-resident immune cells are transported directly across the blood–brain barrier (BBB) and play a significant role in this interaction (M.M.A. et al. 2016). There is evidence suggesting that the gut microbiota influences BBB permeability, as studies have shown that germ-free mice exhibit increased BBB permeability, partly due to reduced expression of tight-junction proteins such as occludin and claudin 5 (Braniste et al. 2014). The microbiota is vital in the initial maturation of the immune system, influencing the expression of toll-like receptors (TLRs) on immune cells and guiding the development of antigen-specific acquired immunity (O’Hara & Shanahan 2006). The hypothesis that alterations in gastrointestinal permeability may trigger systemic inflammatory responses is supported by findings of increased levels of lipopolysaccharides (LPS) and corresponding immunoglobulins in conditions such as depression, autism, and Alzheimer’s disease (Asanka Sanjeewa et al. 2020; Ghosh et al. 2020; Millischer et al. 2021).
Dysbiosis
Dysbiosis refers to the disruption of the delicate balance between the host and its gut microbiota, which can lead to adverse health effects (Levy et al. 2017). Dysbiosis can be triggered by various factors, including antibiotic use, dietary changes, environmental pollutants, and infections, compromising the integrity of the gut barrier, altering immune responses, and resulting in systemic inflammation (Iacob & Iacob 2019; Martinez et al. 2021).
Antibiotic use is a primary driver of dysbiosis, as antibiotics eliminate beneficial microbes, leading to a significant reduction in microbial diversity and the overgrowth of resistant strains (Fröhlich et al. 2016). Dietary changes, particularly those involving high fat and sugar intake, promote the growth of harmful bacteria while reducing beneficial populations, disturbing the microbial balance in the gut (Sonnenburg & Sonnenburg 2019). Environmental pollutants, such as heavy metals and pesticides, can directly affect microbial populations or alter the host’s physiology, further destabilising the gut microbiome (Rinninella et al. 2019). Pathogenic infections outcompete commensal bacteria for resources and niches, triggering inflammatory responses that disrupt the microbial community (C. Wang et al. 2019).
Dysbiosis impairs the interfaces between the host and the microbiome, leading to increased intestinal permeability, immune dysregulation, and systemic inflammation. The disruption of gut barrier integrity often allows harmful substances, including LPS from Gram-negative bacteria, to translocate from the gut lumen into the systemic circulation (Ghosh et al. 2020). LPS, a major component of the outer membrane of Gram-negative bacteria, is a potent activator of the inflammatory response and is recognised by TLRs on immune cells, leading to the production of pro-inflammatory cytokines (Brown 2019).
Dysbiosis can lead to inappropriate immune responses, including increased susceptibility to infections, autoimmune diseases, and chronic inflammatory conditions (Levy et al. 2017). The loss of beneficial microbial populations that produce anti-inflammatory compounds, such as SCFAs, exacerbates immune dysregulation (Sonnenburg & Sonnenburg 2019). The translocation of endotoxins like LPS into the systemic circulation triggers an inflammatory response (explained below). Chronic low-grade inflammation resulting from endotoxemia is associated with various diseases, including metabolic disorders, cardiovascular diseases, and neuroinflammatory conditions (Park et al. 2023).
The inflammatory response to endotoxins significantly impacts multiple pathways. Toll-Like Receptor 4 (TLR4) signalling, for example, recognises LPS and initiates a cascade of immune responses. Upon LPS binding, TLR4 dimerises and recruits adaptor proteins such as Myeloid differentiation primary response 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF), leading to the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the production of pro-inflammatory cytokines, including interleukin-6 (IL-6), tumour necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β) (Asanka Sanjeewa et al. 2020; Hunter & Jones 2015). This pathway is crucial for mounting an effective immune response but can lead to chronic inflammation if dysregulated (Campbell et al. 2014).
The Nucleotide-binding oligomerisation domain, Leucine Rich Repeat and Pyrin domain-containing 3 (NLRP3) inflammasome is a key component in the body’s inflammatory response (Plaza-Díaz et al. 2017). Dysbiosis can activate the NLRP3 inflammasome through microbial products like LPS, leading to the activation of caspase-1 and the maturation of interleukin-1 beta (IL-1β) and interleukin-18 (IL-18). This pathway, while essential for responding to infections, can contribute to inflammatory diseases if overactivated (Guo et al. 2023).
The Janus Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) pathway, activated by pro-inflammatory cytokines like IL-6, also contributes to inflammatory responses. Binding of these cytokines to their receptors leads to the phosphorylation of JAKs, which then phosphorylate STATs. Phosphorylated STATs dimerise and translocate to the nucleus to induce the expression of inflammatory genes, playing a significant role in inflammatory and autoimmune diseases (Asanka Sanjeewa et al. 2020).
The Mitogen-Activated Protein Kinase (MAPK) pathway, another critical signalling mechanism activated by TLRs and cytokine receptors, involves a cascade of protein kinases that lead to the transcription of inflammatory mediators. Dysbiosis-induced activation of the MAPK pathway contributes to the production of cytokines and other inflammatory responses, highlighting its role in chronic inflammation and disease (Sturgeon & Fasano 2016).
Among the pathways activated by dysbiosis-induced inflammation, the KP holds particular significance due to its implications in neurological disorders. The KP is a major route for tryptophan metabolism, producing neuroactive metabolites that significantly impact brain function and fatigue perception. In conditions like Alzheimer’s disease and major depressive disorder, dysregulation of this pathway leads to increased production of neurotoxic metabolites such as quinolinic acid, resulting in neuroinflammation and neurodegeneration (Chatterjee et al. 2018; Marx et al. 2021). This link between the KP and neurological disorders suggests that targeting this pathway may offer therapeutic potential.
Kynurenine Pathway
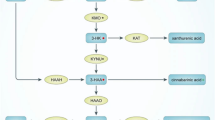
The KP begins with the conversion of tryptophan (TRP) to N-formylkynurenine (N-fKYN), a reaction catalysed by the rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO). N-fKYN is then hydrolysed to kynurenine (KYN) by kynurenine formamidase. TDO primarily facilitates basal TRP metabolism in the liver, whereas IDO is predominantly active in immune cells and can be induced by pro-inflammatory cytokines such as interferon-gamma (IFN-γ), interleukin-1 (IL-1), IL-6, and tumour necrosis factor-alpha (TNF-α) (Fig. 3) (Chen et al. 2021; Hestad et al. 2022).

The tryptophan-kynurenine metabolic pathway. TRP tryptophan; IDO indoleamine 2,3-dioxygenase; TDO tryptophan 2,3-dioxygenase; N-fKYN N-formyl-kynurenine; AA anthranilate acid; KYNU kynureninase; KYN kynurenine; KAT kynurenine aminotransferase; KYNA kynerunic acid; KMO kynurenine 3-monooxygenase; 3-HK 3-hydroxykynurenine; HAAH 3-hydroxyanthranilic acid 3,4-hydroxylase; 3-HAA 3-hydroxyanthranilic acid; HAAO 3-hydroxyanthranilicacid 3,4-dioxygenase; AMS 2-aminomuconic-6-semialdehyde; ACMSD 2-amino-3-carboxymuconate-6-semialdehydedecarboxylase; ACMS 2-amino-3-carboxymuconate-6-semialdehyde; AMSD 2-aminomuconic-6-semialdehyde dehydrogenase; QUIN quinolinic acid; QPRT quinolinate phosphoribosyltransferase; PIC picolinic acid; NAD + nicotinamide adenine dinucleotide
KYN is a crucial metabolite within the pathway, branching into several significant metabolites. One branch converts KYN into kynurenic acid (KYNA) via the enzyme kynurenine aminotransferase (KAT). KYNA is recognised for its neuroprotective properties, acting as an antagonist to the N-methyl-D-aspartate (NMDA) receptor. Another branch converts KYN into anthranilic acid (AA) through the action of kynureninase (KYNU). A third branch transforms KYN into 3-hydroxykynurenine (3-HK) via kynurenine 3-monooxygenase (KMO) (Fig. 3) (Schwartz 2014).
3-HK can be further metabolised into 3-hydroxyanthranilic acid (3-HAA) by KYNU or into xanthurenic acid (XA) by KAT. In the brain, AA is efficiently converted into 3-HAA, which can subsequently form cinnabarinic acid (CA) or 2-amino-3-carboxymuconate-6-semialdehyde (ACMS). ACMS has several metabolic fates: It can be converted to quinolinic acid (QUIN), an NMDA receptor agonist and neurotoxin, or to picolinic acid (PIC) via 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase (ACMSD). QUIN is particularly noteworthy as it can further metabolise into nicotinamide adenine dinucleotide (NAD +) via quinolinate phosphoribosyltransferase (QPRT), highlighting the pathway’s role in cellular energy metabolism (Anderson et al. 2021; Kennedy et al. 2017).
Gut-Derived Inflammation and Its Role in Neuroinflammation
The activation of the KP by pro-inflammatory cytokines links peripheral inflammation to CNS effects. Dysbiosis-induced endotoxemia, through the activation of TLR4 and subsequent cytokine release, can influence the KP. Elevated levels of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β in response to LPS translocation can enhance IDO activity, skewing tryptophan metabolism towards neurotoxic pathways. Furthermore, the metabolites of the KP, particularly QUIN, contribute to the neuroinflammatory milieu. QUIN activates microglia, the resident immune cells of the CNS, further propagating inflammatory responses within the brain. This creates a feedback loop where peripheral inflammation exacerbates central inflammation, potentially leading to or worsening neurological disorders (Calcia et al. 2016; Erickson et al. 2012).
Pathways Leading to Kynurenine Pathway Activation
The inflammatory response induced by endotoxin involves the activation of TLR4 with its co-receptor Myeloid Differentiation factor 2 (MD2) on immune cells. This interaction initiates intracellular signalling cascades involving MyD88, TRAF6, and the IκB kinase (IKK) complex, leading to the activation of NF-κB. NF-κB translocates into the nucleus, upregulating genes encoding pro-inflammatory cytokines such as IL-6 and TNF-α (Lawrence 2009; Nishida et al. 2018; Wu et al. 2018). IL-6 and TNF-α exert their effects by binding to their respective receptors, IL-6R and TNFR1/2, on target cells. IL-6 primarily activates the JAK-STAT pathway, leading to the phosphorylation and activation of STAT3, which regulates inflammatory gene transcription. TNF-α activates various signalling pathways, including NF-κB, Jun N-terminal kinase (JNK), and Mitogen-activated protein kinases (MAPK) pathways. In addition, IFN-γ upregulates the enzyme IDO, catalysing the conversion of tryptophan into N-formylkynurenine, initiating the KP. KMO further converts kynurenine into 3-hydroxykynurenine, an intermediate in the QUIN pathway (Kennedy et al. 2017; Köhler et al. 2021). Pro-inflammatory cytokines like IFN-γ can stimulate KMO, potentially promoting neurotoxicity by favouring the QUIN pathway (Hestad et al. 2022). These inflammatory signals can affect neural drive, contributing to feelings of sadness and increased perceptions of fatigue, potentially leading to neuroinflammatory and neuropsychiatric conditions (Felger 2017; Yamashita 2020). Neuroinflammation mediated by the KP is implicated in the pathogenesis of various neurological disorders, including depression, Alzheimer’s disease, and Parkinson’s disease (Bay-Richter & Wegener 2022; Megur et al. 2021; Venkatesan et al. 2020).
Cytokine Transmission to the Brain
Increased levels of cytokines in the periphery can reach and affect the brain through several mechanisms. These include passage through leaky regions in the BBB such as circumventricular organs, active transport through transport molecules, activation of cells lining the cerebral vasculature (endothelial cells and perivascular macrophages), binding to cytokine receptors associated with the vagus nerve, stimulating the hypothalamic–pituitary–adrenal (HPA) axis at the anterior pituitary or hypothalamus, and recruitment of activated cells such as monocytes/macrophages from the periphery to the brain (Bhatt et al. 2023; Konsman 2022; Kvichansky et al. 2019). Through activation of the intracellular signalling MAPK pathway, cytokines can increase the number and function of the reuptake pumps for serotonin, noradrenaline, and dopamine, which in turn can reduce the availability of these neurotransmitters within the synaptic cleft. Preclinical studies have demonstrated that increased inflammatory cytokines reduce central levels of brain-derived neurotrophic factor (BDNF) and neurogenesis, leading to depressive-like behaviour (Dantzer 2009). However, the relationship between peripheral and central inflammatory markers and antidepressants is complex, and it remains unclear which pathways are most relevant for cytokine signal transmission in stress-related disorders such as depression (Bhatt et al. 2023; Felger 2017; Huang & Wu 2021).
Anti-inflammatory Agents and Depression
There is some evidence, albeit from small studies of short duration, suggesting that anti-inflammatory agents such as non-steroidal anti-inflammatory drugs (NSAIDs) and cytokine inhibitors reduce depressive symptoms (Bhatt et al. 2023). For depressed patients with raised inflammatory markers, this raises the prospect of whether reducing low-grade inflammation could alleviate depressive symptoms. Although a randomised controlled trial of the monoclonal antibody infliximab, a TNF-α antagonist, was not superior to placebo in reducing depressive symptoms overall, in patients with high baseline CRP levels there were greater reductions in depressive symptoms than in those with low CRP levels(Liu et al. 2022). Another study showed that CRP level at baseline differentially predicted treatment outcome with escitalopram or nortriptyline (Fourrier et al. 2018). These studies provide the impetus for stratification of depressed patients based on inflammatory profiles to advance personalised medicine. Developing more nuanced profiles of inflammatory proteins and gene expression, as well as cellular immune parameters, likely represents the future for predictors and targets of response to anti-inflammatory therapies.
Reductions in basal ganglia activity have been noted in more posterior regions, where they are associated with fatigue, and in more ventral regions (such as the nucleus accumbens), where they have been linked to the development of anhedonia(Capuron & Miller 2011; Felger 2017). These areas are crucial for motivation and reward processing, and their impairment can lead to the core symptoms of depression, such as lack of pleasure in activities (anhedonia) and reduced motivation.
The Role of Microglia in Neuroinflammation
Microglia, the resident immune cells of the CNS, are central to the inflammatory process and a source of cytokines. These phagocytic cells account for approximately 10% of cells in the brain and contribute to the plasticity of neural circuits by modulating synaptic architecture and function (Calcia et al. 2016; Erny et al. 2015). Microglial process motility can be modulated by glutamatergic and GABAergic neurotransmission. Acute stress results in microglia activation and increased levels of pro-inflammatory cytokines in areas such as the hippocampus and hypothalamus (Coxon et al. 2018). Most studies show increases in activated microglia in response to chronic stress. Preliminary changes in the microenvironment of the microglia may result in susceptibility to secondary inflammatory stimuli. This concept of microglia priming may be relevant to depression, which often requires multiple environmental “hits” (Setiawan et al. 2015).
In an environmental two-hit rodent model, the first experimental manipulation targeted pregnant dams, and the second manipulation was given to the resulting offspring. Exposure to prenatal immune challenge and peripubertal stress synergistically induced pathological effects on adult behavioural functions and neurochemistry (Giovanoli et al. 2013). Early-life stress primes microglia, leading to a potentiated response to subsequent stress. The microbiota regulates microglia maturation and function, further underscoring the gut–brain axis’s role in neuroinflammation.
Clinically, microglial activation in the prefrontal cortex (PFC), anterior cingulate cortex (ACC), and insula in medication-free depressed patients has been demonstrated using translocator protein density measured by distribution volume in a positron emission tomography study (Deng et al. 2023; Setiawan et al. 2015). This evidence suggests that microglial activation is a significant contributor to neuroinflammation in depression and other neuropsychiatric conditions.
Implications for Neurological Disorders
The chronic activation of the KP can lead to sustained production of neurotoxic metabolites like QUIN, contributing to neurodegeneration and cognitive decline (Hestad et al. 2022). In Alzheimer’s disease, for example, the accumulation of neurotoxic KP metabolites can exacerbate amyloid-beta and tau pathologies, driving disease progression (Guillemin & Brew 2002; Onyango et al. 2021).
In Parkinson’s disease, the dopaminergic neurons in the substantia nigra are particularly vulnerable to KP metabolites, leading to motor symptoms and further neuronal loss (Venkatesan et al. 2020). The role of the KP in these diseases highlights the need for therapeutic strategies targeting this pathway to mitigate neuroinflammation and neurodegeneration.
Therapeutic Potential of Modulating the Kynurenine Pathway
Targeting the KP offers a promising therapeutic strategy for managing neuroinflammatory and neurodegenerative disorders. Inhibitors of IDO and KMO, for instance, can reduce the production of neurotoxic metabolites and shift the balance towards neuroprotective metabolites like kynurenic acid. Preclinical studies have shown that such inhibitors can attenuate neuroinflammation and protect against neuronal damage in models of Alzheimer’s and Parkinson’s diseases (Atlam et al. 2018; Campbell et al. 2014).
Neurological Diseases
Kynurenine Association with Neurological Conditions
Alzheimer’s disease (AD) is characterised by cognitive and memory deficits, amyloid plaques, tau tangles, neuroinflammation, synapse loss, and neuronal loss. Research indicates a significant association between AD and endotoxin levels, with mean blood endotoxin levels in AD patients increased threefold, and brain endotoxin levels elevated two- to threefold. Endotoxin, found within amyloid plaques, can promote amyloid-beta production and aggregation as well as tau hyperphosphorylation. Studies have shown that eliminating gut bacteria reduces plaque load and microglial activation in amyloid model mice (Bello-Medina et al. 2022; K. et al. 2017).
The KP, a major route of tryptophan metabolism, plays a crucial role in AD. Dysregulation of the KP has been observed in AD, leading to increased levels of neurotoxic metabolites and contributing to neuroinflammation and neurodegeneration (Fathi et al. 2022; Guillemin & Brew 2002). Studies have shown altered levels of KP metabolites in the cerebrospinal fluid (CSF) and blood of AD patients, indicating the pathway’s involvement in the disease’s pathogenesis (Si et al. 2023).
Genetic risk for AD is closely linked to APOE isoforms, with APOE2 being protective, APOE3 neutral, and APOE4 detrimental. LPS injection strongly induces serum ApoE in rodents, and ApoE binds LPS, facilitating its uptake and degradation by the liver. APOE4 variant carriers are more sensitive to LPS toxicity than those with APOE3. Variants in the LPS-receptor TLR4 and the LPS-binding receptor TREM2 are also associated with increased AD risk, further establishing the genetic connection between AD and endotoxin (Ghosh et al. 2020; Millischer et al. 2021).
Parkinson’s Disease (PD) is marked by motor dysfunctions, α-synuclein aggregates (Lewy bodies), and dopaminergic neuron loss in the substantia nigra (Kalia & Lang 2015). PD patients often exhibit gut dysfunction and increased gastrointestinal permeability. A significant number of PD patients have elevated blood endotoxin levels, and their gut microbiome differs from that of controls. The gut microbiome’s influence on motor symptoms in PD is evidenced by studies showing that eliminating gut bacteria prevents motor deficits, while introducing the microbiome from PD patients worsens pathology. Colonisation with endotoxin-producing Helicobacter pylori is associated with PD, and its eradication improves symptoms (Lin et al. 2019; Park et al. 2023).
In mice, a single dose of peripheral endotoxin increases α-synuclein expression in intestinal neurons and permeability, mirroring PD patients. Endotoxin also promotes α-synuclein production and fibrillisation. Peripheral endotoxin injection in wild-type and transgenic mice (expressing human A53T mutant α-synuclein) causes acute neuroinflammation, but only transgenic mice develop persistent neuroinflammation, aggregated α-synuclein, and progressive dopaminergic neuron degeneration. This supports the dual-hit hypothesis for PD: endotoxin exposure combined with aggregable α-synuclein leads to neurodegeneration. Nonetheless, endotoxin alone can induce dopaminergic neuron loss in the substantia nigra in mice after several months (Brown 2019; Earls et al. 2019).
The KP is also implicated in PD. Changes in gut microbiota composition have been linked with altered neurotransmitter levels and increased inflammation, potentially contributing to PD progression. The KP of tryptophan catabolism is a key regulator of the immune response and is involved in inflammatory and neurotoxic processes in Parkinson’s disease. Elevated levels of neurotoxic KP metabolites such as QUIN are observed in PD, contributing to neuronal damage and inflammation (Lin et al. 2019; Xiang et al. 2022).
Motor neuron disease, including amyotrophic lateral sclerosis (ALS), involves neurodegeneration of motor neurons and shares genetic and pathological mechanisms with frontotemporal dementia (FTD). Elevated blood endotoxin levels in ALS patients may result from gut inflammation and microbiome alterations. In vitro studies show that LPS addition to microglia or astrocytes causes TDP-43 mis-localisation and aggregation (Quek et al. 2022). Similarly, peripheral LPS administration to TDP-43(A315T) transgenic mice results in TDP-43 aggregation in vivo, suggesting a dual-hit hypothesis for ALS and FTD, where increased endotoxin levels combined with aggregable TDP-43 lead to neurodegeneration(Bright et al. 2021; Correia et al. 2015).
Preventative Measures
The integrity of the intestinal barrier is crucial in preventing the onset and progression of inflammatory diseases. Various strategies have been developed to enhance gut barrier function, addressing both genetic and environmental factors that can compromise intestinal integrity.
Larazotide Acetate is one promising therapeutic agent that inhibits the protein modulator zonulin, which regulates intestinal permeability by modulating TJ disassembly. In coeliac patients, Larazotide Acetate has demonstrated efficacy in reducing disease symptoms by preventing the breakdown of these junctions, thus improving gut barrier function (Leffler et al. 2015). By maintaining the integrity of the intestinal barrier, this drug helps to prevent the translocation of harmful substances, which can trigger systemic inflammation and contribute to various diseases.
Tofacitinib, an inhibitor of inflammatory enzymes associated with rheumatoid arthritis, has shown potential in preventing cytokine-induced gut barrier dysfunction. It works by limiting the delocalisation of TJ proteins such as ZO-1 in intestinal epithelial cells (IECs) exposed to IFN-γ. This action makes it a promising therapeutic agent for inflammatory bowel disease (IBD), where elevated IFN-γ levels in the inflamed intestinal tissues contribute to barrier dysfunction (Spalinger et al. 2021). This drug’s ability to preserve the integrity of the gut barrier highlights its potential in mitigating chronic inflammation associated with IBD (Cleynen et al. 2016).
Another therapeutic approach involves potassium-competitive acid blockers like Tegoprazan, which offer a promising alternative for treating gastric acid-related diseases and protecting against IBD development. Tegoprazan enhances gut barrier health by increasing the abundance of anti-inflammatory bacteria such as Bacteroides vulgatus. These beneficial bacteria help to limit the adhesion of pathogenic bacteria to the epithelial cells, thereby maintaining the integrity of the gut barrier (Son et al. 2022). Unlike proton pump inhibitors (PPIs), which can disrupt the gut microbiota and increase the risk of IBD, Tegoprazan provides a safer option for managing gastric acid without compromising gut health(Kim et al. 2021).
Addressing antibiotic-induced dysbiosis is another critical aspect of maintaining gut barrier integrity. Lentinan, a β-glucan derived from the macrofungus Lentinus edodes, has been shown to counteract the negative effects of antibiotics by boosting beneficial microbial species and promoting the production of short-chain fatty acids (SCFAs) (Zhou et al. 2024). These SCFAs play a vital role in nourishing intestinal epithelial cells and enhancing gut barrier function. In addition, incorporating dietary fibre from sources like the acacia plant before and after antibiotic exposure has been found to reduce E. coli colonisation in animal models. This suggests that specific plant fibres can protect the gut barrier by limiting the growth of pathogenic bacteria (Son et al. 2022).
Dietary strategies are also crucial for enhancing gut barrier integrity. Fermentable dietary fibre, also known as microbiota-associated carbohydrates, promotes the production of SCFAs, which are essential for the health of intestinal epithelial cells. A diet low in fibre has been linked to increased intestinal permeability and a higher abundance of harmful Proteobacteria, highlighting the protective role of dietary fibre (Holscher 2017; Nie et al. 2021). In addition, amino acids like glutamine and arginine, along with minerals such as zinc and vitamins A and D, contribute significantly to gut barrier integrity. Zinc, for example, influences TJ protein expression via the GPR39 receptor, while vitamins A and D modulate the expression of TJ proteins like occludin and ZO-1 ((Den Besten et al. 2013; García-Montero et al. 2021; Xiao et al. 2014; Zuhl et al. 2015).
Probiotics have emerged as a promising intervention for improving intestinal barrier integrity and limiting the development of inflammatory diseases. These live microorganisms, when administered in adequate amounts, confer health benefits on the host by enhancing the gut’s structural and functional integrity (Kaistha & Deshpande 2021). Probiotic strains, primarily from the genera Lactobacillus and Bifidobacterium, are involved in lowering intestinal pH, facilitating cell-to-cell signalling, preventing the colonisation of pathogenic microbes, and regulating the host immune response (Martín & Langella 2019; S. Wang et al. 2021). A subgroup of probiotics, termed “psychobiotics”, has been shown to improve psychological and mental health by influencing mood, anxiety, focus, memory, and cognition (Dinan et al. 2013; Sharma & Bajwa 2022).
Probiotics can modulate gut microbiota composition and restore the gut ecosystem, providing a potential approach for preventing and treating cognitive deficits (Min et al. 2022; Sivamaruthi et al. 2020). For instance, a probiotic mixture of Lactobacillus acidophilus, L. rhamnosus, and Bifidobacteria longum improved symptoms of autism after three months of supplementation (Dargenio et al. 2023; Sivamaruthi et al. 2020). Mechanisms underlying the effects of probiotics on the GBA include modulation of bacterial toxins and amyloid-beta interactions, pathways involving the vagus nerve, cytokines, and neurotransmitters, and impacts on the BBB and intestinal mucosal barrier integrity (Latif et al. 2023; Markowiak & Ślizewska 2017; Nettleton et al. 2021; Rose et al. 2021). Probiotics reduce oxidative stress by producing antioxidant enzymes and substances, such as catalase, superoxide dismutase, butyrate, folate, and glutathione, and chelating metal ions. They may prevent immune responses like inflammation by inhibiting TLR activation and enhancing BBB integrity (X. Wang et al. 2023). In addition, probiotics improve cognitive function in depression by reducing hypothalamic–pituitary–adrenal (HPA) axis dysfunction, increasing monoamine levels, and enhancing neuroplasticity (Ansari et al. 2020; Pinto-Sanchez et al. 2017; Rudzki et al. 2019).
Prolonged probiotic supplementation raises peripheral tryptophan levels, enhancing mental health by upregulating serotonin synthesis enzymes (Mills et al. 2023). Probiotics also modulate neurotransmitters such as serotonin, gamma-aminobutyric acid, acetylcholine, norepinephrine, dopamine, and glutamate, thereby regulating brain activity (Huang & Wu 2021; Lyte 2011; Meeusen & Roelands 2010). Probiotics, such as Bifidobacterium infantis, can restore the normal kynurenine-to-tryptophan ratio, demonstrating the potential of gut microbiota in modulating this pathway and maintaining gut barrier integrity (Purton et al. 2021; Underwood et al. 2015).
Future Directions
The exploration of gut permeability, the GBA, the KP, and their implications for neurological disorders opens numerous avenues for future research. First, there is a need for longitudinal studies to establish causal relationships between gut permeability alterations and the onset of neurological disorders such as Alzheimer’s disease, Parkinson’s disease, and others. Such studies should include diverse populations to account for genetic and environmental variability (Cryan & Dinan 2012).
Further investigation into the molecular mechanisms linking gut permeability to systemic inflammation and neuroinflammation is crucial. This includes identifying specific bacterial strains and their metabolites that influence the KP and tight-junction integrity. Advanced omics technologies, including metagenomics, metabolomics, and proteomics, can provide comprehensive insights into the microbiome–host interactions and their impact on the CNS (O’Mahony et al. 2015).
Clinical trials focusing on therapeutic interventions targeting gut permeability and the KP are essential. Probiotics, prebiotics, and dietary interventions that support gut barrier function should be rigorously tested for their efficacy in preventing or mitigating neurological disorders. In addition, the development of specific inhibitors for enzymes in the KP, such as IDO and KMO, holds promise for reducing neurotoxic metabolite levels and improving neuronal health (Platten et al. 2019).
Investigating the role of the GBA in exercise physiology and its impact on gut permeability and inflammation is another promising area. Understanding how physical activity influences the gut microbiota and the KP could lead to novel strategies for enhancing cognitive function and mental health through exercise (Kennedy et al. 2017).
Lastly, personalised medicine approaches should be explored, considering individual variations in gut microbiota composition and genetic predispositions. Tailoring interventions based on a person’s microbiome profile and KP activity could optimise therapeutic outcomes and minimise adverse effects.
Conclusion
Emerging evidence highlights the potential of targeting gut barrier integrity and the KP as novel therapeutic strategies for preventing and treating conditions such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and other neuropsychiatric disorders. By maintaining a balanced gut microbiota through diet, probiotics, and lifestyle modifications, and by developing specific inhibitors targeting the KP, it may be possible to mitigate neuroinflammation and neurodegeneration.
Data Availability
No datasets were generated or analysed during the current study.
Abbreviations
- AD:
-
Alzheimer’s disease
- CNS:
-
Central nervous system
- EEC:
-
Enteroendocrine cells
- ENS:
-
Enteric nervous system
- GBA:
-
Gut–brain axis
- GI:
-
Gastrointestinal
- HPA:
-
Hypothalamic–pituitary–adrenal
- IA:
-
Intestinal alkaline phosphatase
- IDO:
-
Indoleamine 2,3-dioxygenase
- IEC:
-
Intestinal epithelial cells
- IECs:
-
Intestinal epithelial cells
- IL-1:
-
Interleukin-1
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IL-18:
-
Interleukin-18
- JAM-1:
-
Junctional adhesion molecule 1
- JNK:
-
Jun N-terminal kinase
- KP:
-
Kynurenine pathway
- KAT:
-
Kynurenine aminotransferase
- KMO:
-
Kynurenine 3-monooxygenase
- KYNA:
-
Kynurenic acid
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MS:
-
Multiple sclerosis
- MyD88:
-
Myeloid differentiation primary response 88
- NAD+:
-
Nicotinamide adenine dinucleotide
- NF-Κb:
-
Nuclear factor kappa-light-chain enhancer of activated B cells
- ND:
-
Neurological disorder
- NLRP3:
-
Nucleotide-binding oligomerization domain, Leucine rich repeat and pyrin domain-containing 3
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- PD:
-
Parkinson’s disease
- PPIs:
-
Proton pump inhibitors
- QUIN:
-
Quinolinic acid
- SCFA:
-
Short-chain fatty acids
- STAT:
-
Signal transducer and activator of transcription
- TDO:
-
Tryptophan 2,3-dioxygenase
- TER:
-
Transepithelial resistance
- TJ:
-
Tight junction
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumour necrosis factor-alpha
- TRP:
-
Tryptophan
- ZO-1:
-
Zonula occludens-1
- ZO-2:
-
Zonula occludens-2
- ZO-3:
-
Zonula occludens-3
References
Allam-Ndoul B, Castonguay-Paradis S, Veilleux A (2020) Gut microbiota and intestinal trans-epithelial permeability. Int J Mol Sci. https://doi.org/10.3390/ijms21176402
Almutairi MM, Gong C, Xu YG, Chang Y, Shi H (2016) Factors controlling permeability of the blood–brain barrier. Cell Mol Life Sci 73:57–77
Anderson EW, Fishbein J, Hong J, Roeser J, Furie RA, Aranow C, Volpe BT, Diamond B, Mackay M (2021) Quinolinic acid, a kynurenine/tryptophan pathway metabolite, associates with impaired cognitive test performance in systemic lupus erythematosus. Lupus Sci Med. https://doi.org/10.1136/lupus-2021-000559
Ansari F, Pourjafar H, Tabrizi A, Homayouni A (2020) The effects of probiotics and prebiotics on mental disorders: a review on depression, anxiety, alzheimer, and autism spectrum disorders. Curr Pharm Biotechnol. https://doi.org/10.2174/1389201021666200107113812
Asanka Sanjeewa KK, Nagahawatta DP, Yang HW, Oh JY, Jayawardena TU, Jeon YJ, De Zoysa M, Whang I, Ryu B (2020) Octominin inhibits LPS-induced chemokine and pro-inflammatory cytokine secretion from raw 264.7 macrophages via blocking TLRS/nf-κb signal transduction. Biomolecules. https://doi.org/10.3390/biom10040511
Atlam F, Awad M, Salama R (2018) Factors influencing the potency of alzheimer inhibitors: computational and docking studies. Am J Alzheimer’s Dis Other Demen. https://doi.org/10.1177/1533317517749207
Bailey DM, Evans KA, Mceneny J, Young IS, Hullin DA, James PE, Ogoh S, Ainslie PN, Lucchesi C, Rockenbauer A, Culcasi M, Pietri S (2011) Exercise-induced oxidative-nitrosative stress is associated with impaired dynamic cerebral autoregulation and blood-brain barrier leakage. Exp Physiol. https://doi.org/10.1113/expphysiol.2011.060178
Bay-Richter C, Wegener G (2022) Antidepressant effects of NSAIDs in rodent models of depression—a systematic review. Front Pharmacol. https://doi.org/10.3389/fphar.2022.909981
Bello-Medina PC, Corona-Cervantes K, Zavala Torres NG, González A, Pérez-Morales M, González-Franco DA, Gómez A, García-Mena J, Díaz-Cintra S, Pacheco-López G (2022) Chronic-antibiotics induced gut microbiota dysbiosis rescues memory impairment and reduces β-amyloid aggregation in a preclinical alzheimer’s disease model. Int J Mol Sci. https://doi.org/10.3390/ijms23158209
Berding K, Carbia C, Cryan JF (2021) Going with the grain: fiber, cognition, and the microbiota-gut-brain-axis. Exp Biol Med. https://doi.org/10.1177/1535370221995785
Bhatt S, Devadoss T, Jha NK, Baidya M, Gupta G, Chellappan DK, Singh SK, Dua K (2023) Targeting inflammation: a potential approach for the treatment of depression. Metab Brain Dis. https://doi.org/10.1007/s11011-022-01095-1
Braniste, V., Al-Asmakh, M., Kowal, C., Anuar, F., Abbaspour, A., Tóth, M., Korecka, A., Bakocevic, N., Guan, N. L., Kundu, P., Gulyás, B., Halldin, C., Hultenby, K., Nilsson, H., Hebert, H., Volpe, B. T., Diamond, B., & Pettersson, S. (2014). Erratum: Erratum for the Research Article: The gut microbiota influences blood-brain barrier permeability in mice (Sci. Transl. Med. 6 266er7 (2014)). Science Translational Medicine. https://doi.org/10.1126/scitranslmed.aaa4288
Breit S, Kupferberg A, Rogler G, Hasler G (2018) Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front Psychiatr. https://doi.org/10.3389/fpsyt.2018.00044
Bright F, Chan G, van Hummel A, Ittner LM, Ke YD (2021) Tdp-43 and inflammation: implications for amyotrophic lateral sclerosis and frontotemporal dementia. Int J Mol Sci. https://doi.org/10.3390/ijms22157781
Brown GC (2019) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation. https://doi.org/10.1186/s12974-019-1564-7
Buckley A, Turner JR (2018) Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/cshperspect.a029314
Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD (2016) Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology. https://doi.org/10.1007/s00213-016-4218-9
Campbell BM, Charych E, Lee AW, Möller T (2014) Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci. https://doi.org/10.3389/fnins.2014.00012
Capuron L, Miller AH (2011) Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. https://doi.org/10.1016/j.pharmthera.2011.01.014
Chatterjee P, Goozee K, Lim CK, James I, Shen K, Jacobs KR, Sohrabi HR, Shah T, Asih PR, Dave P, Manyan C, Taddei K, Lovejoy DB, Chung R, Guillemin GJ, Martins RN (2018) Alterations in serum kynurenine pathway metabolites in individuals with high neocortical amyloid-β load: a pilot study. Sci Rep. https://doi.org/10.1038/s41598-018-25968-7
Chen C, Zhang C, Li H, Wang Z, Yuan Y, Zhou M, Fu ZF, Zhao L (2021) Toll-like receptor 4 regulates rabies virus-induced humoral immunity through recruitment of conventional type 2 dendritic cells to lymph organs. J Virol. https://doi.org/10.1128/jvi.00829-21
Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, Brant SR, Cho JH, Daly MJ, Dubinsky M, Duerr RH, Ferguson LR, Franke A, Gearry RB, Goyette P, Lees CW (2016) Inherited determinants of crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. https://doi.org/10.1016/S0140-6736(15)00465-1
Connell CJW, Thompson B, Turuwhenua J, Srzich A, Gant N (2017) Fatigue-related impairments in oculomotor control are prevented by norepinephrine-dopamine reuptake inhibition. Sci Rep. https://doi.org/10.1038/srep42726
Correia AS, Patel P, Dutta K, Julien JP (2015) Inflammation induces TDP-43 mislocalization and aggregation. PLoS ONE. https://doi.org/10.1371/journal.pone.0140248
Coxon JP, Cash RFH, Hendrikse JJ, Rogasch NC, Stavrinos E, Suo C, Yücel M (2018) GABA concentration in sensorimotor cortex following high-intensity exercise and relationship to lactate levels. J Physiol. https://doi.org/10.1113/JP274660
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. https://doi.org/10.1038/nrn3346
Dantzer R (2009) Cytokine, sickness behavior, and depression. Immunol Allerg Clin North Am. https://doi.org/10.1016/j.iac.2009.02.002
Dargenio VN, Dargenio C, Castellaneta S, De Giacomo A, Laguardia M, Schettini F, Francavilla R, Cristofori F (2023) Intestinal barrier dysfunction and microbiota–gut–brain axis: possible implications in the pathogenesis and treatment of autism spectrum disorder. Nutrients. https://doi.org/10.3390/nu15071620
Den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. https://doi.org/10.1194/jlr.R036012
Deng Y, Gong P, Han S, Zhang J, Zhang S, Zhang B, Lin Y, Xu K, Wen G, Liu K (2023) Reduced cerebral cortex thickness is related to overexpression of exosomal miR-146a-5p in medication-free patients with major depressive disorder. Psychol Med. https://doi.org/10.1017/S0033291722003567
Dinan TG, Cryan JF (2017a) Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol. https://doi.org/10.1113/JP273106
Dinan TG, Cryan JF (2017b) The microbiome-gut-brain axis in health and disease. Gastroenterol Clin North Am. https://doi.org/10.1016/j.gtc.2016.09.007
Dinan TG, Stanton C, Cryan JF (2013) Psychobiotics: a novel class of psychotropic. Biol Psychiatr. https://doi.org/10.1016/j.biopsych.2013.05.001
Earls RH, Menees KB, Chung J, Barber J, Gutekunst CA, Hazim MG, Lee JK (2019) Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J Neuroinflammation. https://doi.org/10.1186/s12974-019-1636-8
Erickson MA, Dohi K, Banks WA (2012) Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. NeuroImmunoModulation. https://doi.org/10.1159/000330247
Erny D, De Angelis ALH, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, Schwierzeck V, Utermöhlen O, Chun E, Garrett WS, Mccoy KD, Diefenbach A, Staeheli P, Stecher B, Amit I, Prinz M (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. https://doi.org/10.1038/nn.4030
Fathi M, Vakili K, Yaghoobpoor S, Tavasol A, Jazi K, Hajibeygi R, Shool S, Sodeifian F, Klegeris A, McElhinney A, Tavirani MR, Sayehmiri F (2022) Dynamic changes in metabolites of the kynurenine pathway in alzheimer’s disease, parkinson’s disease, and huntington’s disease: a systematic review and meta-analysis. Front Immunol. https://doi.org/10.3389/fimmu.2022.997240
Felger JC (2017) The role of dopamine in inflammation-associated depression: mechanisms and therapeutic implications. Curr Top Behav Neurosci. https://doi.org/10.1007/7854_2016_13
Fourrier C, Sampson E, Mills NT, Baune BT (2018) Anti-inflammatory treatment of depression: study protocol for a randomised controlled trial of vortioxetine augmented with celecoxib or placebo. Trials. https://doi.org/10.1186/s13063-018-2829-7
Fröhlich EE, Farzi A, Mayerhofer R, Reichmann F, Jačan A, Wagner B, Zinser E, Bordag N, Magnes C, Fröhlich E, Kashofer K, Gorkiewicz G, Holzer P (2016) Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2016.02.020
García-Montero C, Fraile-Martínez O, Gómez-Lahoz AM, Pekarek L, Castellanos AJ, Noguerales-Fraguas F, Coca S, Guijarro LG, García-Honduvilla N, Asúnsolo A, Sanchez-Trujillo L, Lahera G, Bujan J, Monserrat J, Álvarez-Mon M, Álvarez-Mon MA, Ortega MA (2021) Nutritional components in western diet versus mediterranean diet at the gut microbiota-immune system interplay. implications for health and disease. Nutrients. https://doi.org/10.3390/nu13020699
Ghosh SS, Wang J, Yannie PJ, Ghosh S (2020) Intestinal barrier dysfunction, LPS translocation, and disease development. J Endocr Soc. https://doi.org/10.1210/jendso/bvz039
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, Winter C, Riva MA, Mortensen PB, Schedlowski M, Meyer U (2013) Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. https://doi.org/10.1126/science.1228261
Gomez-Eguilaz M, Ramon-Trapero JL, Perez-Martinez L, Blanco JR (2019) [The microbiota-gut-brain axis and its great projections] TT—El eje microbiota-intestino-cerebro y sus grandes proyecciones. Rev Neurol 68(3):111
Guillemin GJ, Brew BJ (2002) Implications of the kynurenine pathway and quinolinic acid in alzheimer’s disease. Redox Rep. https://doi.org/10.1179/135100002125000550
Günzel D, Yu ASL (2013) Claudins and the modulation of tight junction permeability. Physiol Rev 93(2):525–569. https://doi.org/10.1152/physrev.00019.2012
Guo H, Yu L, Tian F, Chen W, Zhai Q (2023) The potential therapeutic role of lactobacillaceae rhamnosus for treatment of inflammatory bowel disease. Foods. https://doi.org/10.3390/foods12040692
Hestad K, Alexander J, Rootwelt H, Aaseth JO (2022) The role of tryptophan dysmetabolism and quinolinic acid in depressive and neurodegenerative diseases. Biomolecules. https://doi.org/10.3390/biom12070998
Holscher HD (2017) Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. https://doi.org/10.1080/19490976.2017.1290756
Huang F, Wu X (2021) Brain neurotransmitter modulation by gut microbiota in anxiety and depression. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2021.649103
Hunter CA, Jones SA (2015) IL-6 as a keystone cytokine in health and disease. Nat Immunol. https://doi.org/10.1038/ni.3153
Iacob S, Iacob DG (2019) Infectious threats, the intestinal barrier, and its trojan horse dysbiosis. Front Microbiol. https://doi.org/10.3389/fmicb.2019.01676
K., E., C., B., F., S., K.H., S., C., P., M., G., & T., H. (2017). Altered gut microbiome composition and digestive enzyme activity of the 5XFAD Alzheimer mouse model. Neurodegenerative Diseases, 17(Supplement 1).
Kaistha SD, Deshpande N (2021) Traditional probiotics, next-generation probiotics and engineered live biotherapeutic products in chronic wound healing. Wound healing research: current trends and future directions. Springer, NY
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet. https://doi.org/10.1016/S0140-6736(14)61393-3
Kennedy PJ, Cryan JF, Dinan TG, Clarke G (2017) Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2016.07.002
Kim SH, Cho KB, Chun HJ, Lee SW, Kwon JG, Lee DH, Kim SG, Jung HY, Kim JW, Lee JS, Park H, Choi SC, Jee SR, Kim HS, Ko KH, Park SJ, Lee YC, Park SH, Kim AR, Song GS (2021) Randomised clinical trial: comparison of tegoprazan and placebo in non-erosive reflux disease. Alimentary Pharmacol Thera. https://doi.org/10.1111/apt.16477
Köhler A, Delbauve S, Smout J, Torres D, Flamand V (2021) Very early-life exposure to microbiota-induced TNF drives the maturation of neonatal pre-cDC1. Gut. https://doi.org/10.1136/gutjnl-2019-319700
Konsman JP (2022) Cytokines in the brain and neuroinflammation: we didn’t starve the fire! Pharmaceuticals. https://doi.org/10.3390/ph15020140
Kvichansky AA, Volobueva MN, Spivak YS, Tretyakova LV, Gulyaeva NV, Bolshakov AP (2019) Expression of mRNAs for IL-1β, IL-6, IL-10, TNFα, CX3CL1, and TGFβ1 cytokines in the brain tissues: assessment of contribution of blood cells with and without perfusion. Biochem Mosc 84:905–910
Latif A, Shehzad A, Niazi S, Zahid A, Ashraf W, Iqbal MW, Rehman A, Riaz T, Aadil RM, Khan IM, Özogul F, Rocha JM, Esatbeyoglu T, Korma SA (2023) Probiotics: mechanism of action, health benefits and their application in food industries. Front Microbiol. https://doi.org/10.3389/fmicb.2023.1216674
Laurent F, Hillery AM, Mrsny R (2016) Overview of epithelial barriers. In: Drug delivery: Fundamentals and applications, 2nd edn. https://doi.org/10.1201/9781315382579
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/cshperspect.a001651
Lee B, Moon KM, Kim CY (2018) Tight junction in the intestinal epithelium: its association with diseases and regulation by phytochemicals. J Immunol Res. https://doi.org/10.1155/2018/2645465
Leffler DA, Kelly CP, Green PHR, Fedorak RN, Dimarino A, Perrow W, Rasmussen H, Wang C, Bercik P, Bachir NM, Murray JA (2015) Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology. https://doi.org/10.1053/j.gastro.2015.02.008
Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E (2017) Dysbiosis and the immune system. Nat Rev Immunol. https://doi.org/10.1038/nri.2017.7
Lin CH, Chen CC, Chiang HL, Liou JM, Chang CM, Lu TP, Chuang EY, Tai YC, Cheng C, Lin HY, Wu MS (2019) Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J Neuroinflammation. https://doi.org/10.1186/s12974-019-1528-y
Liu H, Wu X, Wang Y, Liu X, Peng D, Wu Y, Chen J, Su Y, Xu J, Ma X, Li Y, Shi J, Yang X, Rong H, Forti MDi, Fang Y (2022) TNF-α, IL-6 and hsCRP in patients with melancholic, atypical and anxious depression: an antibody array analysis related to somatic symptoms. Gen Psych. https://doi.org/10.1136/gpsych-2022-100844
Louwies T, Johnson AC, Orock A, Yuan T, Greenwood-Van Meerveld B (2020) The microbiota-gut-brain axis: an emerging role for the epigenome. Exp Biol Med. https://doi.org/10.1177/1535370219891690
Lyte M (2011) Probiotics function mechanistically as delivery vehicles for neuroactive compounds: microbial endocrinology in the design and use of probiotics. BioEssays. https://doi.org/10.1002/bies.201100024
Madara JL (1998) Regulation of the movement of solutes across tight junctions. Annu Rev Physiol 60:143–159. https://doi.org/10.1146/annurev.physiol.60.1.143
Markowiak P, Ślizewska K (2017) Effects of probiotics, prebiotics, and synbiotics on human health. Nutrients. https://doi.org/10.3390/nu9091021
Martín R, Langella P (2019) Emerging health concepts in the probiotics field: streamlining the definitions. Front Microbiol. https://doi.org/10.3389/fmicb.2019.01047
Martinez JE, Kahana DD, Ghuman S, Wilson HP, Wilson J, Kim SCJ, Lagishetty V, Jacobs JP, Sinha-Hikim AP, Friedman TC (2021) Unhealthy lifestyle and gut dysbiosis: a better understanding of the effects of poor diet and nicotine on the intestinal microbiome. Front Endocrinol. https://doi.org/10.3389/fendo.2021.667066
Marx W, McGuinness AJ, Rocks T, Ruusunen A, Cleminson J, Walker AJ, Gomes-da-Costa S, Lane M, Sanches M, Diaz AP, Tseng PT, Lin PY, Berk M, Clarke G, O’Neil A, Jacka F, Stubbs B, Carvalho AF, Quevedo J, Fernandes BS (2021) The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: a meta-analysis of 101 studies. Mol Psych. https://doi.org/10.1038/s41380-020-00951-9
Matsuda M, Kubo A, Furuse M, Tsukita S (2004) A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci. https://doi.org/10.1242/jcs.00972
Meeusen R, Roelands B (2010) Central fatigue and neurotransmitters, can thermoregulation be manipulated? Scand J Med Sci Sports. https://doi.org/10.1111/j.1600-0838.2010.01205.x
Megur A, Baltriukienė D, Bukelskienė V, Burokas A (2021) The microbiota–gut–brain axis and alzheimer’s disease: neuroinflammation is to blame? Nutrients. https://doi.org/10.3390/nu13010037
Millischer V, Heinzl M, Faka A, Resl M, Trepci A, Klammer C, Egger M, Dieplinger B, Clodi M, Schwieler L (2021) Intravenous administration of LPS activates the kynurenine pathway in healthy male human subjects: a prospective placebo-controlled cross-over trial. J Neuroinflammation. https://doi.org/10.1186/s12974-021-02196-x
Mills S, Yang B, Smith GJ, Stanton C, Ross RP (2023) Efficacy of Bifidobacterium longum alone or in multi-strain probiotic formulations during early life and beyond. Gut Microbes. https://doi.org/10.1080/19490976.2023.2186098
Min S, Than N, Shin YC, Hu G, Shin W, Ambrosini YM, Kim HJ (2022) Live probiotic bacteria administered in a pathomimetic Leaky Gut Chip ameliorate impaired epithelial barrier and mucosal inflammation. Sci Rep. https://doi.org/10.1038/s41598-022-27300-w
Mor A, Tankiewicz-Kwedlo A, Krupa A, Pawlak D (2021) Role of kynurenine pathway in oxidative stress during neurodegenerative disorders. Cells. https://doi.org/10.3390/cells10071603
Morris MC, Gilliam EA, Li L (2015) Innate immune programing by endotoxin and its pathological consequences. Front Immunoly. https://doi.org/10.3389/fimmu.2014.00680
Nettleton JE, Klancic T, Schick A, Choo AC, Cheng N, Shearer J, Borgland SL, Rho JM, Reimer RA (2021) Prebiotic, probiotic, and synbiotic consumption alter behavioral variables and intestinal permeability and microbiota in btbr mice. Microorganisms. https://doi.org/10.3390/microorganisms9091833
Nie Q, Hu J, Gao H, Li M, Sun Y, Chen H, Zuo S, Fang Q, Huang X, Yin J, Nie S (2021) Bioactive dietary fibers selectively promote gut microbiota to exert antidiabetic effects. J Agric Food Chem. https://doi.org/10.1021/acs.jafc.1c01465
Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A (2018) Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. https://doi.org/10.1007/s12328-017-0813-5
O’Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep. https://doi.org/10.1038/sj.embor.7400731
O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF (2015) Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. https://doi.org/10.1016/j.bbr.2014.07.027
Odenwald MA, Turner JR (2017) The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. https://doi.org/10.1038/nrgastro.2016.169
Okumura R, Takeda K (2017) Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med. https://doi.org/10.1038/emm.2017.20
Onyango IG, Jauregui GV, Čarná M, Bennett JP, Stokin GB (2021) Neuroinflammation in alzheimer’s disease. Biomedicines. https://doi.org/10.3390/biomedicines9050524
Park JM, Lee SC, Ham C, Kim YW (2023) Effect of probiotic supplementation on gastrointestinal motility, inflammation, motor, non-motor symptoms and mental health in Parkinson’s disease: a meta-analysis of randomized controlled trials. Gut Pathog. https://doi.org/10.1186/s13099-023-00536-1
Pinto-Sanchez MI, Hall GB, Ghajar K, Nardelli A, Bolino C, Lau JT, Martin FP, Cominetti O, Welsh C, Rieder A, Traynor J, Gregory C, De Palma G, Pigrau M, Ford AC, Macri J, Berger B, Bergonzelli G, Surette MG, Bercik P (2017) Probiotic bifidobacterium longum NCC3001 reduces depression scores and alters brain activity: a pilot study in patients with irritable bowel syndrome. Gastroenterology. https://doi.org/10.1053/j.gastro.2017.05.003
Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. https://doi.org/10.1038/s41573-019-0016-5
Plaza-Díaz J, Ruiz-Ojeda FJ, Vilchez-Padial LM, Gil A (2017) Evidence of the anti-inflammatory effects of probiotics and synbiotics in intestinal chronic diseases. Nutrients. https://doi.org/10.3390/nu9060555
Purton T, Staskova L, Lane MM, Dawson SL, West M, Firth J, Clarke G, Cryan JF, Berk M, O’Neil A, Dean O, Hadi A, Honan C, Marx W (2021) Prebiotic and probiotic supplementation and the tryptophan-kynurenine pathway: a systematic review and meta analysis. Neurosci Biobehav Rev. https://doi.org/10.1016/j.neubiorev.2020.12.026
Quek H, Cuní-López C, Stewart R, Colletti T, Notaro A, Nguyen TH, Sun Y, Guo CC, Lupton MK, Roberts TL, Lim YC, Oikari LE, La Bella V, White AR (2022) ALS monocyte-derived microglia-like cells reveal cytoplasmic TDP-43 accumulation, DNA damage, and cell-specific impairment of phagocytosis associated with disease progression. J Neuroinflammation. https://doi.org/10.1186/s12974-022-02421-1
Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC (2019) What is the healthy gut microbiota composition? a changing ecosystem across age, environment, diet, and diseases. Microorganisms. https://doi.org/10.3390/microorganisms7010014
Rose EC, Odle J, Blikslager AT, Ziegler AL (2021) Probiotics, prebiotics and epithelial tight junctions a: promising approach to modulate intestinal barrier function. Int J Mol Sci. https://doi.org/10.3390/ijms22136729
Rudzki L, Ostrowska L, Pawlak D, Małus A, Pawlak K, Waszkiewicz N, Szulc A (2019) Probiotic Lactobacillus Plantarum 299v decreases kynurenine concentration and improves cognitive functions in patients with major depression: a double-blind, randomized, placebo controlled study. Psychoneuroendocrinology. https://doi.org/10.1016/j.psyneuen.2018.10.010
Sarkar A, Lehto SM, Harty S, Dinan TG, Cryan JF, Burnet PWJ (2016) Psychobiotics and the manipulation of bacteria–gut–brain signals. Trends Neurosci. https://doi.org/10.1016/j.tins.2016.09.002
Schoultz I, Keita ÅV (2020) The intestinal barrier and current techniques for the assessment of gut permeability. Cells. https://doi.org/10.3390/cells9081909
Schwartz CE (2014) Aberrant tryptophan metabolism: The unifying biochemical basis for autism spectrum disorders? Biomarkers Med. https://doi.org/10.2217/bmm.14.11
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, Suridjan I, Kennedy JL, Rekkas PV, Houle S, Meyer JH (2015) Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatr. https://doi.org/10.1001/jamapsychiatry.2014.2427
Sharma H, Bajwa J (2022) Approach of probiotics in mental health as a psychobiotics. Arch Microbiol. https://doi.org/10.1007/s00203-021-02622-x
Si ZZ, Zou CJ, Mei X, Li XF, Luo H, Shen Y, Hu J, Li XX, Wu L, Liu Y (2023) Targeting neuroinflammation in alzheimer’s disease: from mechanisms to clinical applications. Neural Regen Res. https://doi.org/10.4103/1673-5374.353484
Sivamaruthi BS, Suganthy N, Kesika P, Chaiyasut C (2020) The role of microbiome, dietary supplements, and probiotics in autism spectrum disorder. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph17082647
Son M, Park IS, Kim S, Ma HW, Kim JH, Kim TI, Kim WH, Han J, Kim SW, Cheon JH (2022) Novel potassium-competitive acid blocker, tegoprazan, protects against colitis by improving gut barrier function. Front Immunol 13:870817
Sonnenburg ED, Sonnenburg JL (2019) The ancestral and industrialized gut microbiota and implications for human health. Nat Rev Microbiol. https://doi.org/10.1038/s41579-019-0191-8
Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, Penna G, Dejana E, Rescigno M (2015) A gut-vascular barrier controls the systemic dissemination of bacteria. Science. https://doi.org/10.1126/science.aad0135
Spalinger MR, Sayoc-Becerra A, Ordookhanian C, Canale V, Santos AN, King SJ, Krishnan M, Nair MG, Scharl M, Mccole DF (2021) The JAK inhibitor tofacitinib rescues intestinal barrier defects caused by disrupted epithelial-macrophage interactions. J Crohn’s Colitis. https://doi.org/10.1093/ecco-jcc/jjaa182
Sturgeon C, Fasano A (2016) Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers. https://doi.org/10.1080/21688370.2016.1251384
Suzuki T (2020) Regulation of the intestinal barrier by nutrients: the role of tight junctions. Animal Sci J. https://doi.org/10.1111/asj.13357
Takiishi T, Fenero CIM, Câmara NOS (2017) Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. https://doi.org/10.1080/21688370.2017.1373208
Tlaskalová-Hogenová H, Štěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, Rossmann P, Hrnčíř T, Kverka M, Zákostelská Z, Klimešová K (2011) The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 8(2):110–120
Underwood MA, German JB, Lebrilla CB, Mills DA (2015) Bifidobacterium longum subspecies infantis: champion colonizer of the infant gut. Pediatr Res. https://doi.org/10.1038/pr.2014.156
Ursell LK, Metcalf JL, Parfrey LW, Knight R (2012) Defining the human microbiome. Nutr Rev. https://doi.org/10.1111/j.1753-4887.2012.00493.x
Venkatesan D, Iyer M, Narayanasamy A, Siva K, Vellingiri B (2020) Kynurenine pathway in parkinson’s disease—an update. eNeurologicalSci. https://doi.org/10.1016/j.ensci.2020.100270
Wang C, Li Q, Ren J (2019) Microbiota-immune interaction in the pathogenesis of gut-derived infection. Front Immunol. https://doi.org/10.3389/fimmu.2019.01873
Wang S, Wang B, Mishra S, Jain S, Ding J, Krtichevsky S, Kitzman D, Yadav H (2021) A novel probiotics therapy for aging-related leaky gut and inflammation. Innov Aging. https://doi.org/10.1093/geroni/igab046.2521
Wang X, Wang Z, Cao J, Dong Y, Chen Y (2023) Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome. https://doi.org/10.1186/s40168-022-01452-3
Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, Theodorou V, Dekker J, Méheust A, De Vos WM, Mercenier A, Nauta A, Garcia-Rodenas CL (2017) Homeostasis of the gut barrier and potential biomarkers. Am J Physiol—Gastrointest Liver Physiol. https://doi.org/10.1152/ajpgi.00048.2015
Wu J, Ding J, Yang J, Guo X, Zheng Y (2018) MicroRNA roles in the nuclear factor kappa B signaling pathway in cancer. Front Immunol. https://doi.org/10.3389/fimmu.2018.00546
Xiang S, Ji JL, Li S, Cao XP, Xu W, Tan L, Tan CC (2022) Efficacy and safety of probiotics for the treatment of alzheimer’s disease, mild cognitive impairment, and parkinson’s disease: a systematic review and meta-analysis. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2022.730036
Xiao S, Fei N, Pang X, Shen J, Wang L, Zhang B, Zhang M, Zhang X, Zhang C, Li M, Sun L, Xue Z, Wang J, Feng J, Yan F, Zhao N, Liu J, Long W, Zhao L (2014) A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol. https://doi.org/10.1111/1574-6941.12228
Yamashita M (2020) Potential role of neuroactive tryptophan metabolites in central fatigue: establishment of the fatigue circuit. Int J Tryptophan Res. https://doi.org/10.1177/1178646920936279
Zhou G, Liu H, Yuan Y, Wang Q, Wang L, Wu J (2024) Lentinan progress in inflammatory diseases and tumor diseases. Eur J Med Res. https://doi.org/10.1186/s40001-023-01585-7
Zhu H, Wang W, Li Y (2024) The interplay between microbiota and brain-gut axis in epilepsy treatment. Front Pharmacol. https://doi.org/10.3389/fphar.2024.1276551
Zuhl M, Dokladny K, Mermier C, Schneider S, Salgado R, Moseley P (2015) The effects of acute oral glutamine supplementation on exercise-induced gastrointestinal permeability and heat shock protein expression in peripheral blood mononuclear cells. Cell Stress Chaperones. https://doi.org/10.1007/s12192-014-0528-1
Acknowledgements
The authors would like to thank Ulster University for agreeing to this work.
Funding
Ulster University.
Author information
Authors and Affiliations
Contributions
R.K was the only author.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Ethical Approval
This is a review study. No ethical approval required.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kearns, R. Gut–Brain Axis and Neuroinflammation: The Role of Gut Permeability and the Kynurenine Pathway in Neurological Disorders. Cell Mol Neurobiol 44, 64 (2024). https://doi.org/10.1007/s10571-024-01496-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10571-024-01496-z