
Abstract
A possibility of using aluminum hydroxide, a product of processing of the adsorbent that lost its sorption activity in purification of a polyethylene solution cyclohexane, was demonstrated in the synthesis of aluminum kaolin borate supports for hydrodesulfurization catalysis in oil refining. The influence of typical impurities V, Ti and Na in the composition of the spent adsorbent on the physicochemical and catalytic properties of supports and catalysts was determined. Alumina-kaolin-borate catalyst carriers with the Ti content of approx. 0.2% have been prepared by atomic layer deposition (ALD) of TiCl4 onto the granules. A positive effect of molecular layering with TiO2 has been revealed by temperature programmed reduction and oxidation in combination with hydrodesulphurization activity testing.
Graphical Abstract

Similar content being viewed by others
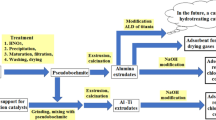

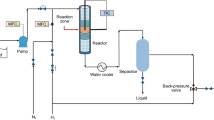
Avoid common mistakes on your manuscript.
1 Introduction
As a part of the global efforts related to circular economy, a significant attention is given to recycling of spent catalysts. Hydrodesulfurization is one of the key processes in oil refining, being typically carried out in the presence of Ni-Mo, Ni-W and Co-Mo sulfide catalysts, supported on refractory oxides such as γ-alumina under with hydrogen pressure and temperature varying between 0.5 and 4 and even 8 MPa and 300–400 °C respectively depending on the feedstock [1,2,3]. Oxide catalyst precursors, in the form of trilobe or pentalobe extrudates or hollow cylinders with diameters of 1.5–2 mm and lengths of 3–5 mm, are preliminarily subjected to sulfidation.
In industry, Al-Co-Mo and Al-Ni-Mo hydrodesulfurization catalysts are most widely used [4], when desulfurization of various feedstock with simultaneous removal of nitrogen and oxygen is required along with dearomatization [1,2,3,4,5,6]. There is a substantial amount of literature available on the influence of preparation methods [7], catalyst composition, type of the support (e.g. alumina, titania, zirconia, silica) [8], reducibility of the metal [9], its dispersion, structure of the active sites in the oxide and sulfided forms [7,8,9,10,11,12,13,14].
The mechanism of hydrodesulfurization on sulfided Al-Co-Mo and Al-Ni-Mo was studied using various model substances such as thiophene, benzothiophene [15], and dimethyldibenzothiophene [7], emphasizing differences in the catalytic HDS behavior of cobalt and nickel. Using toluene as an example, a correlation was established between the hydrogenating ability and the Co/Mo ratio in crystallites, and the decisive role of mixed Co-Mo centers decorating “CoMoS” nanocrystals in the sulfided catalysts was noted [16].
In addition to the influence of operation parameters (temperature and flow rate [14]), presence of various additives including compounds containing phosphorus [11, 12] and boron [13, 17] was demonstrated to affect hydrodesulfurization of real oil fractions. Moreover, as already indicated acidity of Al2O3-TiO2 and Al2O3-MgO carriers [14] can also have an impact. Of large importance for improving hydroprocessing catalysts is the uniform distribution of the active phase precursors and diminishing interactions with the support surface [18], as strong interactions of the hydrogenating metals (Ni, Co, Mo) with γ-Al2O3 lead to lower amounts of the MoS2 phase active in hydrodesulfurization [19].
In the context of circular economy and utilization of waste, it is tempting to apply alumina-containing industrial waste for preparation of environment-friendly hydrodesulfurization catalysts suitable in HDS of oil refining streams. One of such hazardous wastes generated in large quantities, is spent alumina adsorbent (SAA) applied for purification of the waste cyclohexane solution, i.e. the reaction medium for polymerization of ethylene into polyethylene with organometallic compounds as catalysts. After completion of operation, SAA contained deposits of deactivated polymerization catalysts organometallic compounds of VC16O5H14 and TiC16O4H14, as well as chlorine-containing substances. When processing it into a fresh adsorbent for purifying exhaust gases from a naphtha reformer, SAA granules were successively washed with an excess of 1% NaOH solution, deionized water and calcined at 400° C [20]. This is followed by activation in 8–9% NaOH solution at 40 °C, with subsequent drying at 150–180 °C.
Previously, the authors have considered utilization of the spent alumina adsorbent by processing SAA into fresh pseudo-boehmite, which was then applied for synthesis of sorbents for removal of chlorinated compounds [20, 21]. The authors made some preliminary attempts to use the alumina-containing industrial waste SAA for preparation of environment-friendly hydrodesulfurization catalysts suitable in HDS of oil refining streams [20, 22,23,24].
Based on these studies, it was concluded that impurities of vanadium and titanium compounds were not completely removed when treating SAA with an alkaline solution [23]. On the other hand, presence of titania in combination with a more traditional alumina support has been demonstrated for catalytic transformations relevant to biomass valorization via hydrodeoxygenation [25].
The main aim of this work was assessment of utilization of spent alumina adsorbents as a carrier for NiMo catalysts exploring especially the role of titania introduced to the support by atomic layer deposition (ALD). ALD is a method based on the sequential use of a gas-phase chemical process, in particular alternating reactions of TiCl4 with surface hydroxyl groups of Al2O3 and hydrolysis, leading to deposition of a thin film. The resulting catalysts were tested in a pilot unit with real refinery feeds.
2 Experimental
2.1 Materials
Details of the preparation of the supports have been reported previously [20]. Here the most pertinent details are given. The waste aluminium oxide adsorbent (SAA) was supplied from the Shurtan gas chemical complex, and had the chemical composition: and while kaolin was procured from the Angren quarry—"Angren Kaolin" LLC.
To modify the adsorbents, the following materials were used: boric acid 99.6% H3BO3 (Ekotec) and TiCl4 99.9% – SKT Taurus.
The following materials have been used for modification of adsorbents: NaOH from AO Navoiyazot, and Na3VO4.12H2O, chemically pure, from Chem Craft. Other reagents for preparation of the hydrotreating catalysts are listed below: (NH4)4Mo7O24 × 4H2O—(Uzbek Factory of Refractory and Heat Resistant Metals, Chirchik), phosphoric acid 98.5% H3PO4 (Ammofos OJSC, Almalyk), Ni(NO3)2 × 6H2O and Co(NO3)2 × 6H2O (JSC Ural Factory of Chemical Reagents).
2.2 Synthesis of Catalysts
Waste processing consisted of preliminary dissolution of crushed SAA by heating in a solution of nitric acid with the formation of aluminum nitrate, precipitation of AlOOH by adding an ammonia solution at pH = 7–8, followed by maturation of the precipitate at 20–27 °C, filtering and washing, and finally drying the powder pseudoboehmite AlOOH. The grinded SAA (30% by weight with the particle size between 0.04 and 0.1 mm) and kaolin (10% by weight) were added to AlOOH powder during preparation of the carrier, named sample S1akb, when peptization of the mixture of dry ingredients was carried out by stepwise addition of 3 wt% nitric acid and 2.5 wt.% boric acid solutions. The peptized paste was extruded into trefoil-shaped granules with a circumferential diameter of 4 mm and a length of 3–5 mm. After drying in air, the S1akb granules were calcined for three hours at 550 °C. A sample of the AKB-Ti(0.18) support (hereinafter S2ald) was obtained by applying TiCl4 vapor to S1akb granules using the ALD method [20]. Here and further in the sample designations, the number in brackets after the symbol of a chemical element corresponds to its content in wt%. The degree of ALD was monitored by absorption bands of surface hydroxyl groups in the near-infrared (NIR) spectrum at 4500 and 7300 cm–1, which decreased due to interaction with TiCl4. In the IR spectrum of S1akb dehydrated at 100 °C, a set of narrow bands 3745; 3730; 3710; 3552 cm–1 from OH groups was associated with aluminum, silicon and boron atoms, along with a wide band in the region of 3333–3400 cm–1 from incompletely removed H2O molecules. Subsequently Ti–OH bonds were formed upon exposure to water vapours and hydrolysis of Ti-Cl bonds followed by exposure again to dry nitrogen at 200 °C to remove the excess of water vapours. Formation of Ti–OH bonds was accompanied by an increase in the intensity of the bands at 4500 and 7300 cm–1 in the NIR spectrum, as well as at 3670 and 3720 cm–1 in the IR spectrum. The ALD cycles have been repeated to deposit the desired layers of titania. Formation of the titania layer on the surface of the granules was controlled by appearance in the Raman spectrum of an intense band at ca. 142 cm– 1. The intense band at 142 cm–1, along with low-intensity bands at 196, 396, 512 and 639 cm–1, is characteristic of the crystalline anatase phase. The band in the region of 142 cm−1 appeared in the Raman spectra of TiO2 nanopowders with the nanocluster sizes of 5 to 30 nm [26] not being present in the spectrum of the original S1akb carrier.
The Ni(3.1)Mo(8.3)P(0.9)/AKB-Ti(0.18) catalyst (hereinafter referred to as C-1ald) was prepared by single impregnation of the trilobes of the S2ald support with a combined aqueous solution of (NH4)4Mo7O24 × 4H2O and Ni(NO3)2 × 6H2O, stabilized with phosphoric acid, according to moisture absorption. Ni(3.5)Mo(11.2)P(1.6)/AKB and Ni(0.7)Co(2.5)Mo(8.2)P(0.9)/AKB-Ti(0.18) catalysts (hereinafter referred to as C-2akb and C-3ald) were obtained by impregnating the supports: first with an aqueous solution of (NH4)4Mo7O24 × 4H2O, stabilized by an appropriate amount of orthophosphoric acid, and after heat treatment at 300 °C with an aqueous solution of nickel nitrate or a mixture of nickel and cobalt nitrates. Ni(3.8)Mo(11.0)P(0.35)/AKB, Ni(4.4)Mo(14.4)P(3.1)/AKB and Co(3.1)Mo(8.2)P(0.26)/AKB-Ti(0.18) catalysts (hereinafter referred to as C-4akb, C-5akb and C-6ald) before applying aqueous solutions of nickel or cobalt nitrate were pre-impregnated twice with an aqueous solution of (NH4)4Mo7O24 × 4H2O, stabilized with H3PO4 with an intermediate heat treatment at 300 °C. As a result, after the final heat treatment at 500 °C, the materials presented in Table 1 were obtained. For comparison purpose, Table 1 shows also characteristics of the industrial catalyst Co(3.4)Mo(8.83)P(0.3)/Al2O3 designated Cind.
The limited liquid uptake by the supports required the use of concentrated solutions to obtain catalysts of the calculated composition. Stable solutions of (NH4)4Mo7O24 × 4H2O were obtained in the pH range = 1.3 ÷ 1.5, when, in addition to hydrated MoO42− ions, [NiH6Mo6O24]4– heteropolyanions were formed, which coexisted with anions of the [CoH6Mo6O24]4– type, NO3– and PO4– 3 anions and cations Co2+, Ni2+, NH4+ [22]. The choice of H3PO4 as a reagent to prevent precipitation in the impregnating solutions was related to its impact on the structure of active phases namely formation of heteropolycompounds of transition metals [23].
The elemental analysis of the samples has been performed with scanning electron microscope (SEM) EVOMA 10 combined with EDX. The total content of titanium, vanadium, nickel and molybdenum in the obtained samples was also determined by photocolorimetry and of sodium by the flame photometry. The textural properties have been determined by mercury porosimetry (Carlo Erba) operating up 200 MPa.
Mechanical strength was determined by cutting the granule crosswise using a device with a special knife. The breaking load was calculated according to:
where P is the breaking load, kg; m – the cargo weight, kg; l1 – distance from the beginning of the scale to the center of the load, cm; l2 is the distance from the beginning of the scale to the middle of the knife, cm. To compare the catalysts, the specific strength value was used calculated as the average value of the breaking load for 30 measurements divided by the granule diameter (mm).
XRD was done using Empyrean diffractometer. Assignment of the reflexes in the diffraction patterns was carried out using JCPDS card file. Crystallite sizes were calculated using the Scherrer equation:
where D is the average crystallite/ grain size in the powder (nm), n is a coefficient depending on the shape of the particles (n is taken equal to 0.9, similar to [7, 27]), λ = 1.54 is the radiation wavelength (Cu), β is the physical broadening of the line in the diffraction pattern in radians (on a scale of 2 θ).
Electronic diffuse reflectance spectra (ESDR) of the catalysts were recorded in the region of 50–10 kcm −1, on a Hitachi-330 spectrophotometer. The samples were first ground to a powder state, then pressed in a special cuvette followed by recording the spectra using MgO as a standard. Potential distortion of the spectra by species containing Ni, Co, Mo due to the support influence was taken into account [22]. The data are presented below using the Kubelka–Munk function F(R∞),
where R∞ is the reflectance at infinite depth [11].
The strength and concentration of acid–base centers on the surface of the samples calcined at 350 °C without contact with air were determined from the electronic spectra of adsorbed indicators [22]. Indicators, characterized by their respective pKa value in parentheses below, triphenylcarbinol (-6.3), benzalacetophenone (-5.6), dicinthalacetone (-3.3), benzolazo diphenylamine (+ 1.5) and bromophenol blue (+ 3.8), exhibited specific absorption bands in the visible ESDR region upon interactions with surface aprotic and proton centers of varying strengths. The appearance in the ESDR spectrum of adsorbed triphenycarbinol of a band at 22.7–23.2 kcm−1 and a band at 19.0 kcm−1 in the spectrum of adsorbed dicinthalacetone indicated their adsorption on strong acidic sites of the Brønsted type (protic acidity). The presence of a band at 21.0 kcm−1 in the spectrum of adsorbed dicinthalacetone indicated the presence of Lewis-type centers (aprotic acidity). To determine the concentration of surface centers with a given pKa value, two spectra were obtained. The first of them characterized the state of the molecules of the corresponding indicator directly on the surface of the original sample, while the second corresponded to the spectrum of the indicator on the surface after poisoning the acid sites with a given amount of n-butylamine. The concentration of acid sites was determined by:
where Cacid – concentration of acid sites in mmol/g, Cbut – concentration of butylamine in mmol /g, F(R∞) and F(R∞)but are Kubelka–Munk functions at the maximum of the band of the acid form of the corresponding indicator, respectively, before and after poisoning of the surface acid sites with butylamine.
Raman spectra were obtained on an InVia Raman spectrometer (Renishaw). The measurements were carried out at room temperature. A Cobalt CW 532 nm DPSS laser RL532C100 (diode-pumped solid state laser) with a wavelength of 532 nm and a nominal energy of 100 mW was used as an excitation source.
Extraction of soluble substances from the catalysts was carried out first with distilled water and then with a 12.5 M aqueous ammonia solution. The extraction procedure was carried out three times with intermediate calcination at 500 °C. The content of the elements Co, Ni, Mo, Ti P, Si and Al extracted from the catalysts with water or an aqueous ammonia solution was determined by absorption spectroscopy using an SP 9 atomic adsorption spectrophotometer PYE UNICAM.
The temperature programmed reduction was performed with Auto Chem 2910 HP system, equipped with the thermal conductivity detector. First, a sample (100 mg) was heated in the quartz reactor from the ambient temperature to 500 °C at the rate of 5 °C/min under the nitrogen flow rate of 50 ml/min, followed by holding at this temperature for 30 min. After cooling to the room temperature in the nitrogen flow, 10 vol.% of hydrogen (20 ml/min) in argon was used for the catalyst reduction increasing temperature with the ramp 10 °C/min.
Prior to oxygen chemisorption, the catalyst sample (1 g) was heated to 500 °C under 10 vol.% of hydrogen in argon following by holding at the highest temperature for 30 min. Upon cooling to 30 °C, oxygen pulses on 0.2 ml were conducted recording the amounts of unreacted oxygen at the reactor outlet.
The catalysts were tested in hydrotreating of three types of hydrocarbon feedstock:
-
1)
naphtha—crude gasoline fraction with a boiling point (Tboil) of 30–90° C, including mainly n-paraffins, iso-paraffins C3-C6 and naphthenes, as well as traces of benzene derivatives C6-C7, along with olefins (< 1%) and sulfur containing compounds (Sini) – 0.15%;
-
2)
crude kerosene fraction with the boiling point Tboil. of 120—315 °C, including mainly n-paraffins, iso-paraffins C3-C9 of ca. 38% and naphthenes—about 25%, polyaromatic hydrocarbons C3-C9 (PAH)—18%, as well as impurities of benzene derivatives C6-C8 and olefins—no more than 1%, and finally sulphur containing compounds with Sinitial—0.25%, including mercaptans—0.02%;
-
3)
deasphalted residue with Tboil. above 500–590 °C, containing predominantly paraffin-naphthenic hydrocarbons, PAH—33.1%.
Among impurities the following should be mentioned: resinous-asphaltene substances 1.27%, sulfur (Sinitial) -1.32% and nitrogen—0.1%. The iodine value, which reflects the amount of unsaturated hydrocarbons, was 0.5 g I2/100 g. Activity of the catalysts in hydrodesulfurization and hydrogenation of polyaromatic compounds in the fed was evaluated in a continuous fixed-bed reactor (Scheme 1) under conditions typical for industry. After loading into the reactor, the catalysts were sulfided at 400 °C for 4 h in a flow of 10 vol. % H2S in H2 under atmospheric pressure [23].

Schematic of a high-pressure pilot plant for testing activity of hydrotreating catalysts. 1 – burette; 2 – piston pump; 3 – hydrogen cylinder; 4 – pressure gauge; 5 – reactor; 6—catalyst bed; 7 – porcelain balls; 8 – thermocouple; 9 – temperature regulator; 10-reactor heating; 11- cooler; 12- high pressure separator; 13 – low pressure separator; 14 –sample receiver; 15 – H2S absorber; 16 – gas meter
Testing of catalytic properties was carried out at a volumetric feed rate of 2 h−1, hydrogen to hydrocarbon feedstock ratio of 300 m3/m3, 320 °C and pressure (MPa): 3.0 for naphtha, 4.0 for crude kerosene fraction and 6.0 MPa for deasphalted residue. The change in the content of polyaromatic compounds was assessed based on the results of structural group analysis using the R-D-M method (refractive index-density-molecular weight).
3 Results and Discussion
3.1 Texture and Surface Properties of Materials
SEM analysis of the elemental composition of S1akb and S2ald carriers revealed mainly the presence of Al, Si and O (Fig. 1 on the left), and impurity elements (V, Ti, Cl and others) were detected only at individual points. Titanium was unambiguously detected only when scanning the surface of granules of the S2ald support or the near-surface layer (Fig. 1 in the center). When scanning the surface of the catalyst granules, Ti practically did not appear in the spectra (Fig. 1 on the right). The average results of SEM analysis of transition elements and phosphorus at arbitrary points in the volume and on surface of the catalysts, as well as physicochemical characteristics are presented in Table 1.

SEM analysis of the S2ald carrier in the main body of the granules (left) and in the near-surface layer (center); catalyst Ni(0.7)Co(2.5)Mo(8.2)P(0.9)/AKB-Ti(0.18) – on the right
As can be seen from Table 1, introduction of a catalytically active phase decreased the surface area of S1akb supports, while for S2ald only minor changes were observed.
The mechanical crushing strength along the diameter of the granules decreased slightly after ALD modification of the carrier with titanium dioxide.
A decrease in the mechanical strength and the specific surface area after impregnation of the catalytically active phase occurred due to a decrease in the diameter of small pores in the final catalysts. Mo, P, Co, Ni as well as Al containing compounds were deposited in the pores due to partial leaching of the support in the impregnating solutions [28]. The numbers in the parentheses after the symbol of an element indicate its concentration in weight percent. No aprotic acid sites were found on the surface of all the supports and catalysts studied, in contrast to CoMo/γ-Al2O3P(2)Ce(1.75) catalysts [12]. ALD modification with titania was accompanied by a redistribution of acid sites on the surface of the S1akb support. Rather strong proton centers with pKa <—5.6 disappeared at the expense of the concentration of weaker acid sites. Brønsted acid sites were found on the surface of the catalysts, the concentration of which increases with increasing concentrations of molybdenum and phosphorus, decreasing with increasing molar ratio Co + Ni/Mo.
3.2 XRD
The diffraction pattern of the S2ald carrier, described in [20] and presented in Fig. 2, did not illustrate specific TiO2 reflections corresponding to the interplanar distances d = 3.52 Å (anatase) or d = 3.25 and 2.487 Å (rutile) due to the low concentration and high dispersion of deposited titanium dioxide using the ALD method. According to SEM data [20], titanium was located in the outer layer, while diffraction patterns were taken in the form of a compressed powder of crushed S2ald granules. Only broad peaks from γ-Al2O3 and narrow peaks from α-SiO2 were observed, as in the diffraction pattern of the unmodified S1akb support. Wide peaks from kaolinite with d = 7.17; 4.46; 3.57 and 2.48 Ǻ, characteristic of Angren kaolin, did not appear in the studied samples due to its destruction during calcination.

Diffractograms: 1)S2ald, 2) C-2akb, 3) C-4akb; 4) C-5akb; 5) C-1ald, 6) C-6ald; 7) C-3ald; 8) Cind (industrial catalyst).
 – γ—Al2O3;
– γ—Al2O3;
 – SiO2;
– SiO2;
 – CoMoO4;
– CoMoO4;
 – NiMoO4;
– NiMoO4;
 – MoO3
– MoO3
Similar diffraction patterns were obtained on a titanium-containing support for C-3ald and C-1ald materials. Intense peaks of SiO2 and broad peaks of γ-Al2O3 partially or completely overlapped the main peaks, corresponding to probable compounds: MoO3, NiO, CoO, Co3O4, NiAl2O4, CoAl2O4, NiMoO4 and CoMoO4. This made interpretation of the diffraction patterns challenging, but could indicate a high dispersion of hydrogenating metal compounds in the catalysts supported on S2ald with a moderate amount of phosphorus introduced during impregnation [29]. The diffraction patterns of the synthesized nickel molybdenum catalysts differed significantly from the catalyst (1 mmol Ni, 3.1 mmol Mo per 1 g Al2O3) obtained by impregnation of alumina with an aqueous solution of both nickel hexahydrate and ammonium molybdate tetrahydrate, where rather narrow peaks of NiMoO4 and NiAl2O4 were present, as well as a weak broad NiO peak [6].
Calculations using the Scherrer equation (peak 2 θ = 28.52°) for an industrial sample of Cind, not containing SiO2 and TiO2, with similar amounts of Co, Mo and P in the composition C-6ald, revealed similar sizes of CoMoO4 crystallites – 3.55 nm and 3.05 nm, respectively. The size of NiMoO4 crystallites (peak 2 θ = 29.2°) in the C-4akb catalyst was of the same order—3.21 nm. The NiMoO4 crystallite sizes increased slightly as the phosphorus content increased above 1%, up to 3.33 nm in the C-2akb catalyst. The sizes of Co(Ni)MoO4 crystallites coincided with the average particle sizes of about 3.1 nm in calcined catalysts of the composition Co(2.89–3.71)Mo(11.95)P(0.86–4.45) on γ-Al2O3, determined by transmission electron microscopy (TEM) [16]. At higher concentrations of nickel, molybdenum and phosphorus in the C-5akb catalyst, the calculated sizes of NiMoO4 crystallites already reached 14.5 nm. The appearance of a single peak 2 θ = 23.4° from MoO3 [9] with particle sizes of 7.17 and 58.6 nm in the catalysts containing more than 1% phosphorus indicated poor dispersion of molybdenum oxide. A slight increase in the background in the angle range 2 θ = 10.0–13.80° was probably due to the presence of Ni polymolybdate, including [Mo7O24]−6 ions [7].
3.3 ESDR
The ESDR of the pressed powders of the S1akb and S2ald carriers were identical; only very weak absorption bands were observed at 510 and 550 nm from the Fe3+-O-Fe2+ impurity in the kaolin composition [22]. In ESDR of whole S2ald granules, obvious absorption in the region of 200–345 nm was attributed to the O–2 → Ti4+ charge transfer band characteristic of anatase [14, 27]. In the ESDR of catalysts obtained in the “Auto zero” mode relative to the corresponding supports (Fig. 3), absorption bands were observed indicating different coordination of transition metal ions, described in detail in the literature [7, 14, 22,23,24]. A strong charge transfer band at 200–220 nm is attributed to the superposition of absorption bands from isolated MoO42− particles with the tetrahedral coordination Mo6+Td and polymolybdates of the MoTd6+ – O – MoOh6+ type. The shift of the edge of this band to the long-wavelength region, observed at higher content of molybdenum and phosphorus, indicated formation of the polymer chains MoOh6+ – O – MoOh6+, as well as the bulk phase MoO3 in the C-5akb and C-2akb catalysts [9]. The spectral manifestation of cobalt ions in octahedral coordination (Co Oh2+) included bands in the region of 469 and 510 nm. Bulk NiMoO4 with the octahedral coordination – Ni Oh2+ corresponded to a system of bands at 417–435 and 735–813 nm, while weak bands at 714 and 769 nm indicated the presence of surface nickel-molybdate structures, and bands in the region of 322 and 333 nm are attributed to the surface associates of the Al-O-Ni–O-Mo- type. A doublet of bands at 595–602 and 628–633 nm from Ni Td2+ and a triplet at 555, 602 and 666 nm from Co Td2+ indicated partial incorporation of group VIII ions into tetrahedral vacancies of alumina. Absence of non-stoichiometric oxides NiO1+x and CoO1+x in the catalyst composition follows from a low absorption level in the region of 330–1000 nm in the ESDR spectra.

ESDR of catalysts: 1) C-2akb; 2) C-4akb; 3) C-5akb; 4) C-1ald; 5) C-6ald; 6) C-3ald; 7) Cind. A) -catalysts calcined after extraction with water, b) calcined after extraction with ammonia solution
3.4 Raman Spectra
Raman spectroscopy is widely used to identify various transition metal compounds that are formed during synthesis of hydrotreating catalysts, depending on the characteristics of their preparation [29]. In the Raman spectra of the S1akb and S2ald carriers, weak bands from Al2O3 appeared at 250 cm−1 (Fig. 4) [31]. The anatase form of TiO2 in the S2ald spectrum corresponded to a clear band of moderate intensity at 141.4 cm-1 and weak bands at 397, 520 and 638 cm−1 [26, 27]. In the spectra of catalysts on S2ald, titanium dioxide appeared only as a band at 141.4–142.0 cm−1, since weak high-frequency bands overlapped with bands from molybdenum compounds, in contrast to the NiMo catalyst on a support containing 95% TiO2 [14]. The set of bands 966–967, 940, 890, 926, 590, 370 and 230 cm−1 in the Raman spectra of impregnating solutions of (NH4)4Mo7O24×4H2O stabilized H3PO4 (pH of 1.25–1.54) is better corresponding to the spectrum of the Mo8O264− [7] and P2Mo5O234− anions rather than Mo7O246− [8, 29].

Raman spectra: 1) S1akb; 2) S2ald; 3) C-2akb; 4) C-4akb; 5) C-5akb; 6) C-1ald; 7) C-6ald; 8) C-3ald; 9) Cind
The Raman spectra of the catalysts contained the main bands from polymolybdate ions in the high-frequency region 880–1023 cm−1, the intensity ratio and the position of the maxima of which varied depending on the concentration of Mo and P, as well as on the Mo/P ratio (Fig. 4). The MoO42− bands in the region of 910–916 cm−1, comparable in intensity to the bands of polymolybdates, were observed for catalysts containing no more than 8.3 wt.%. molybdenum (spectra 6–8 in Fig. 4). The bands at 890–1000 cm−1 and 830–970 cm−1 in the Raman spectra were attributed to symmetric and asymmetric stretching vibrations of the terminal Mo = O bond. The band around 310–370 cm−1 corresponded to the bending vibrations of the Mo = O terminal bond, and the bands around 560 and 210 cm−1 were assigned to the symmetric Mo–O-Mo stretching vibrations and the Mo–O-Mo bending vibrations, respectively. The intensity of the band of polymolybdate anions with a characteristic peak in the region of 570–580 cm−1 [19, 29] was at maximum in the Raman spectrum of the C-5akb catalyst and decreased as the concentration of molybdenum and phosphorus decreased (Fig. 4).
The complex shape of intense bands with peaks in the region of 912–1010 cm–1 of catalysts on S2ald support is due to the superposition of a number of signals from polymolybdates, normal molybdates and molybdenum oxide. Presence of MoO42− ions in the Raman spectra in Fig. 4 was evidenced by groups of bands close to those published in the literature for NiMoO4 species (709 and 964 cm–1) and Al2(MoO4)3 (~ 993, 1004, 1026 cm–1 – bending vibrations of the Mo = O bond of three MoO4 units and 822, 888, 915 cm–1 – stretching vibrations of the Mo = O bond of three MoO4 units) [6].
Lower intensity and blurriness of the characteristic band of polymolybdate compounds in the Raman spectra (from vibrations of the terminal groups M = O) (Fig. 4) was observed in the range of 954–965 cm−1. This along with the subtle bands also corresponding to the crystalline phases of NiMoO4 (915 and 720 cm−1) and MoO3 (1000–993, 820, 670 cm−1) [7, 29], indicated higher dispersion of transition metals in C-1ald compared to C-2akb.
A wide band from 840 to 940 cm−1, attributed in [7] to tetrahedral MoO42− ions present in NiMoO4 and CoMoO4 phases, was masked by intense bands of polymolybdate ions (950 cm−1) and large MoO3 crystals (820 cm−1), identified by XRD (Fig. 2).
Low-intensity bands at about 373 cm−1 in the spectra of the C-2akb, C-4akb, C-5akb catalysts and 196–198 cm−1 of the Cind catalyst are attributed to the low crystalline structures of NiAl2O4 and CoAl2O4, respectively [31], which were absent in the case of catalysts based on S2ald. Formation of Co3O4 and NiO was not detected. Specific lines of Co3O4 (472, 514,610 and 678 cm−1) and NiO (540 cm−1) clearly appeared in the Raman spectra of oxides, obtained by precipitation from the corresponding nitrates with a NaOH solution.
3.5 Selective Extraction of Active Components
Table 2 presents the data for the elements in aqueous and aqueous-ammonia extracts from the catalysts, as well as the carrier. As mentioned in [29], during extraction with water highly dispersed MoO3 and transition metal molybdates species are removed from hydrotreating catalysts, being just weakly bound to the surface. During the subsequent alkaline hydrolysis, when under the influence of NH4OH, -Al-O–Mo, -Al-O–Mo-O-Ni- and similar bonds were destroyed, the structures most active in hydrodesulfurization reactions migrated into the aqueous ammonia solution. The maximum amount of structures weakly associated with the S1akb support was 7.2% for Mo, and 2.56% for Ni of the weight of the initial C-2akb catalyst. On the ALD modified support, the amount of structures extracted with water from the C-6ald catalyst reached 6.3% for Mo ca. 2.7% for Co. The minimum amount of water-soluble compounds was recorded for the industrial catalyst: Mo – 3.73%, and Co –1.72%. A large number of structures extracted with an aqueous ammonia solution were present in the C-5akb catalyst: 4.94% Mo, as well as in industrial Cind catalyst: 4.12% Mo. The presence of a certain amount of Ti, Si, P and Al in the extracts may indicate their interactions with molybdenum polyanions in the acidic environment of the impregnation solution with the formation of Al2(MoO4)3 and heteropoly compounds. These elements were practically absent in the extracts of the blank experiment with the S2ald carrier. Raman spectra (Fig. 4) confirmed the presence of Al2(MoO4)3 [6] in the catalysts under study. The spectroscopic data contradict possible preservation of Mo and P complexes during calcination, in particular: P2Mo5O235− (956, 926, 870 cm−1); HP2Mo5O235− (936, 882 cm−1); H2P2Mo5O234− (944, 894 cm−1); H2PMo11CoO405− (1008, 971, 954, 886, 816 cm−1), described in [18].
3.6 TPR
Analysis of the profile of TPR curves of oxide forms of catalysts makes it possible to estimate the strength of the metal-support interactions and predict the transformations of specific structures reduced by hydrogen at low temperatures into catalytically active Co(Ni)MoS type II phases during sulfidation prior to hydrotreating [12]. The obtained data coincided with the published data [24, 30]. Under the selected conditions, the TPR curve of Co3O4 had a peak at 340 °C with a small shoulder at about 400 °C. The reduction of NiO occurred at 300 °C, while transformation NiTd2+ → Nio in the NiAl2O4 was initiated above 700 °C. The S1akb and S2ald carriers did not absorb hydrogen within the experimental sensitivity. Sulfidation of catalysts in hydrogen environment, as well as removal of sulfur, nitrogen and oxygen from hydrocarbon feedstocks, is carried out at temperatures rarely exceeding 400 °C [4, 7, 11, 14,15,16,17, 19], thus position of the first peak in the TPR curves can be considered as the most relevant for predicting catalytic properties. Consumption of hydrogen for the reduction of oxides present in the catalysts based on the S2ald carrier was detected at 250 – 300 °C (Fig. 5, curves 2–4).

The TPR profiles: 1)C-2akb; 2) C-6ald; 3) C-1ald; 4) C-3ald; 5) C-4akb; 6) C-5akb; 7) Cind
The maximum hydrogen consumption due to the reduction of octahedrally coordinated ions NiOh2+ (CoOh2+) and MoOh6+ → Mo+5 in the structure of the corresponding polymolybdates and dispersed phase MoO3 was observed in the range of 380–400 °C [7, 24]. The first maximum in the TPR curve of the C-4akb catalyst of a similar composition on the S1akb support was observed at 405 °C (Fig. 5, curve 5) and shifted to 430 °C with an increase in the phosphorus content from 0.35 to 1.6% at a similar concentration of active metals. The second maximum in the TPR curves in the region of 480–540 °C was attributed to the reduction of NiOh2+(CoOh2+) in the structure of the corresponding monomolybdates with a simultaneous intensification of the reactions MoOh6+ → Mo+5 → Mo+4. The intensive involvement of NiTd2+ and CoTd2+ ions in the spinel structures, as well as MoTd6+ in monomolybdates, with a possible transition to the metallic state, into the reduction process was evidenced by high-temperature maxima in the TPR curves in the region of 685–725 °C with a wide shoulder from the continuing hydrogen consumption. The diffraction patterns of the samples, taken after completion of the TPR experiment at 900 °C, contained peaks corresponding to the metal particles Nio and Coo. Peaks from Moo were not detected, since complete restoration of molybdate structures required aging the samples in a hydrogen environment at a temperature of at least 915 °C [28].
Calculation of the area under the TPR curves, reflecting the amount of consumed hydrogen in the temperature range 300–500 °C at the first stage of reduction of polymolybdate structures, NiMoO4 and CoMoO4, revealed the maximum hydrogen consumption of ca. 2.1 mmol H2/g for C-4akb and C-3ald catalysts (Table 3). Although the multi-peak nature of the TPR curves (Fig. 5) did not allow an accurate determination of hydrogen consumption for reduction of a particular phase, the temperature of the first maximum, along with the total hydrogen consumption in the temperature range typical for the operation of hydrotreating catalysts, made it possible to compare the easiness of hydrogenating elements reduction.
Considering that sulfidation of oxide forms of hydrotreating catalysts is carried out in hydrogen environment with sulfur-containing compounds at 320–400 °C [12, 14], and hydroprocessing is carried out at 260–400 °C [7, 15, 17], hydrogen consumption for TPR per catalyst weight was calculated for two temperatures of two temperature intervals 250–400 °C and 300–500 °C (Table 3). The amount of oxygen chemisorbed by equal amounts of catalysts pre-treated with hydrogen at 500 °C indirectly indicated dispersion of the reduced metals. C-4akb catalyst with a NiMoO4 crystal size of about 3.2 nm, differing from C-2akb not only in the amount of phosphorus, but also in larger MoO3 crystals absorbed ca. 1.4 fold more oxygen, which can be attributed to difference in the dispersion of oxides. The negative effect of excess phosphorus was even more pronounced for the C-5akb catalyst. Reduced catalysts on the S2ald support with a lower content of hydrogenating elements absorbed more oxygen, which correlated well with the results of estimating the crystal sizes of MoO3, CoMoO4 and NiMoO4 using the Scherrer formula.
3.7 Catalyst Activity
A comparison of the hydrodesulfurization function of catalysts in the hydrotreating of hydrocarbon fractions that differ substantially in the sulfur content and the boiling points (Table 4) revealed high activity of the ALD modified samples. Based on the results of 30 days tests in hydrotreating of deasphalted residue and kerosene, it was shown that stability of the C-3ald catalyst at the same level as unmodified C-4akb. Co-Mo and Co–Ni-Mo catalysts with molybdate crystal sizes smaller than 4 nm, containing less than 0.9 wt.% phosphorus and having Brønsted acid sites of medium strength on the surface, removed sulfur with a very similar efficiency from naphtha (> 99.8%), kerosene (> 94%) and deasphalted residue (66.67–72.7%). The C-4akb catalyst, en par in terms of desulfurization activity with the industrial catalyst, had a better dearomatization performance. The C-4akb catalyst combined with the demetallization catalyst ZSh-25 (Uzbekistan) and GO-70 (Russia), was in operation at the Fergana Oil Refinery for 6 years providing a required decrease in the concentration of polyaromatic hydrocarbons in the production of base oils from deasphalted vacuum distillation residue.
4 Conclusions
The influence of ALD modification of an aluminum kaolin borate carrier obtained from industrial waste on the physicochemical characteristics of hydrodesulfurization catalysts was studied using XRD, ESDR, Raman spectroscopy as well as TPR. Conditions for the preparation of C-4akb catalysts, which combine a high hydrodesulfurization function with activity in the hydrogenation of polyaromatic hydrocarbons, have been optimized. The results of determining the activity at the pilot plant revealed certain advantages of the molecular layering of titanium dioxide on the aluminum kaolin borate carrier S1akb in the processes of hydrodesulfurization of hydrocarbon raw materials, especially mercaptan sulfur—an important parameter in the production of aviation fuel.
References
Topsøe H (1996) Clausen BS and Massoth FE Hydrotreating catalysis. Springer, Berlin
Moulijn JA, Makee M, Van Diepen AE (2013) Chemical process technology, 2d Edition, Weinheim, Wiley
Murzin DYu (2022) Chemical reaction technology, 2nd Edition, Berlin, De Gruyter
Petrova D, Lyubimenko V, Ivanov E, Gushchin P, Kolesnikov I (2022) Energy basics of catalytic hydrodesulfurization of diesel fuels. Catalysts 12:1301
Ding S, Jiang S, Wang J, Huang X, Yang Z (2020) Effects of the Ni-Mo ratio on olefin selective hydrogenation catalyzed on Ni-Mo-S active sites: A theoretical study by DFT calculation. Fuel 277:118136
Kumar P, Maity SK, Shee D (2019) Role of NiMo alloy and Ni species in the performance of NiMo/alumina catalysts for hydrodeoxygenation of stearic acid: A kinetic study. ACS Omega 4:2833–2843
Badoga S, Ganesan A, Dalai AK, Chand S (2016) Effect of synthesis technique on the activity of CoNiMo tri-metallic catalyst for hydrotreating of heavy gas oil. Catal Today 291:160–171
Hu H, Wachs IE (1995) Surface structures of supported molybdenum oxide catalysts: characterization by Raman and Mo L3-Edge XANES. J Phys Chem 99:10897–10910
Priecel P, Kubicka D, Vázquez-Zavala A, de los Reyes AJ, Pouzar M, Capek L (2020) Alternative preparation of improved NiMo-alumina deoxygenation catalysts. Front Chem 8:216
Zhang D, Liu X-M, Liu Y-X, Yan Z-F (2020) Impact of γ-alumina pore structure on structure and performance of Ni–Mo/γ-Al2O3 catalyst for 4,6-dimethyldibenzothiophene desulfurization. Microporous Mesoporous Mater 310:110637
de Mello MD, de Almeida BF, da Costa MB, Zotin JL, da Silva MAP (2018) Kinetic modeling of deep hydrodesulfurization of dibenzothiophenes on NiMo/alumina catalysts modified by phosphorus. Fuel Process Technol 177:66–74
Xia B, Cao L, Luo K, Zhao L, Wang X, Gao J, Xu C (2019) Effects of the active phase of CoMo/γ-Al2O3 catalysts modified using cerium and phosphorus on the HDS performance for FCC gasoline. Energy Fuels 33:4462–4473
Rinaldi N, Al-Dalama K, Kubota T, Okamoto Y (2009) Preparation of Co–Mo/B2O3/Al2O3 catalysts for hydrodesulfurization: Effect of citric acid addition. Applied Catalysis A 360:130–136
Vazquez-Garrido I, Lopez-Benitez A, Berhault G, Guevara-Lara A (2019) Effect of support on the acidity of NiMo/Al2O3-MgO and NiMo/TiO2-Al2O3 catalysts and on the resulting competitive hydrodesulfurization/hydrodenitrogenation reactions. Fuel 236:55–64
Shafiq I, Shafique S, Akhter P, Yang W, Hussain M (2020) Recent developments in alumina supported hydrodesulfurization catalysts for the production of sulfur-free refinery products: A technical review. Catal Rev: Sci Eng 64:1–86
Costa V, Guichard B, Digne M, Legens C, Lecour P, Marchand K, Raybaud P, Krebs E, Geantet C (2013) A rational interpretation of improved catalytic performances of additive-impregnated dried CoMo hydrotreating catalysts: a combined theoretical and experimental study. Catal Sci Technol 3:140–151
Wu G, Yin Y, Chen W, Xin F, Lu Y, Qin K, Zhang L, Song Y, Li M (2019) Catalytic kinetics for ultra-deep hydrodesulfurization of diesel. Chem Eng Sci 214:115446
Catita L, Quoineaud A-A, Moreaud M, Espinat D, Pichon C, Delpoux O (2018) Impact of citric acid on the impregnation of CoMoP/γ-Al2O3 catalysts: time and spatially resolved MRI and Raman Imaging study. Top Catal 61:1474–1484
Hu D, Li H-P, Mei J-L, Xiao C-K, Wang E-H, Chen X-Y, Zhang W-X, Duan A-J (2022) The effect of chelating agent on hydrodesulfurization reaction of ordered mesoporous alumina supported NiMo catalysts. Pet Sci 19:321–328
Yunusov MP, Gulomov ShT, Nasullaev KhA, Turdiyeva DPK, Isaeva NF, Abdurakhmanova ISK, Mustafaev BD, Murzin DYu (2024) Mitigating the environmental impact by synthesis of adsorbents from aluminium-containing waste. Waste Biomass Valori. https://doi.org/10.1007/s12649-023-02387-y
Yunusov MP, Nasullaev KhA, Gulomov ShT, İsaeva NF, Mustafaev BD, Rakhimjanov BB, Khodjiev RG (2020) Analysis of the results of experimental sorbent for chloride compounds removal. Chem Probl 3:366–375
Yunusov MP, Saidaxmedov ShM, Djalalova ShB, Nasullaev KhA, Gulyamov ShT, Isaeva NF, Mirzaeva EI (2015) Synthesis and research of Co-Ni-Mo catalysts of oil fractions hydroprocessing. Catal Sustain Energy 2:43–56
Nasullaev KhA, Gulomov ShT, Sayidov UKh, Teshabaev ZA, Yunusov MP (2017) Experimental evidences of the direct influence of composition and prepared conditions on activity and selectivity of hydrotreating catalysts. Chem Probl 2:173–179
Yunusov MP, Djalalova ShB, Nasullaev KhA, Gulyamov ShT, Mirzaeva EI, Teshabaev ZA (2013) Study of the influence of titanium-containing carriers on the reducibility and dispersibility of nickel and molybdenum structures. Uzbek J Oil Gas 3:47–49
Valdés-Martínez OU, Santolalla-Vargas CE, Santes V, de los Reyes JA, Pawelec B, Fierro JLG (2019) Influence of calcinations on metallic dispersion and support interactions for NiRu/TiO2 catalyst in the hydrodeoxygenation of phenol. Catal Today 329:149–155
Mamedov S (2020) Characterization of TiO2 nanopowders by Raman spectroscopy. Spectrosc Raman Technol Today’s Spectroscopists 35:41–49
Gowthambabu V, Govindaraj MDR, Krishna VKN, Charumathi ML, Kumar JM, Vignesh MSD, Daniel RI, Ramasamy P (2021) Synthesis of anatase TiO2 microspheres and their efficient performance in dye-sensitized solar cell. J Mater Sci: Mater Electron 32:26306–26317
Ferdous D, Dalai AK, Adjaye J (2004) A series of NiMo/Al2O3 catalysts containing boron and phosphorus. Part I. Synthesis and characterization. Appl Catal A 260:137–151
Chukin GD (2010) The structure of aluminum oxide and hydrodesulfurization catalysts. Reaction mechanisms. Moscow, p 287
Carrier X, Marceau E, Che M (2006) Physical techniques and catalyst preparation: Determining the interactions of transition-metal complexes with oxide surfaces. Pure Appl Chem 78:1039–1055
Horsth DFL, Primo JO, Balaba N, Anaissi FJ, Bittencourt C (2023) Color stability of blue aluminates obtained from recycling and applied as pigments. RSC Sustain 1:159–166
Funding
Open access funding provided by Åbo Akademi University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yunusov, M.P., Gulomov, S., Nasullayev, K.A. et al. Hydrodesulfurization Catalysts from Aluminium-Containing Waste. Catal Lett (2024). https://doi.org/10.1007/s10562-024-04685-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10562-024-04685-z