
Abstract
Introduction
Homologous recombination (HR) is a crucial DNA-repair mechanism, and its disruption can lead to the accumulation of mutations that initiate and promote cancer formation. The key HR genes, BRCA1 and BRCA2, are particularly significant as their germline pathogenic variants are associated with a hereditary predisposition to breast and/or ovarian cancer.
Materials and methods
The study was performed on 45 FFPE breast cancer tissues obtained from 24 and 21 patients, with and without the germline BRCA1/2 mutation, respectively. The expression of 11 genes: BRCA1, BRCA2, ATM, BARD1, FANCA, FANCB, FANCI, RAD50, RAD51D, BRIP1, and CHEK2 was assessed using Custom RT2 PCR Array (Qiagen), and results were analysed using R.
Results
Cancer tissues from patients with BRCA1 or BRCA2 germline mutations displayed no significant differences in the expression of the selected HR genes compared to BRCA1 or BRCA2 wild-type cancer tissues. In BRCA1mut cancer tissues, BRCA1 expression was significantly higher than in BRCA2mut and BRCA wild-type cancer tissues.
Conclusions
In cancer tissues harbouring either BRCA1 or BRCA2 germline mutations, no significant differences in expression were observed at the mRNA level of any tested HR genes, except BRCA1. However, the significant differences observed in BRCA1 expression between germline BRCA1mut, germline BRCA2mut and BRCA1/2wt tissues may indicate a compensatory mechanism at the mRNA level to mitigate the loss of BRCA1 function in the cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) is a highly heterogeneous disease with many different histological, molecular, and clinical subtypes. The molecular heterogeneity is observed at the genetic, epigenetic, and transcriptomic levels. This, combined with the high level of morbidity, makes BC an interesting subject of scientific studies focused on understanding the causes and mechanisms of BC development and progression [1,2,3].
Each BC is unique and differs at the molecular level. Thus, the classification of BC based on histological and immunohistochemical features does not reflect the complex genetic changes underlying tumour growth and development [4].
Gene expression analyses allow for identifying molecular BC subtypes, thus, enabling tailored therapy. Moreover, currently, available multigene prognostic panels, such as Oncotype Dx and MammaPrint, rely on the expression of various genes in the tumour tissue and, thus, differ in the range of prognostic and/or predictive capabilities. Therefore, developing new tests based on wide genetic analyses and introducing them into clinical practice are a promising area in the diagnosis and treatment of this cancer [5, 6].
Maintaining genetic stability is extremely difficult and is enabled by constantly active DNA-repair systems [7, 8]. An accumulation of DNA mutations resulting from ineffective DNA repair leads to genome instability, a key reason for cancer development and progression. Therefore, mutations in DNA-repair genes are frequent targets for tailored therapies [8, 9].
It has been revealed that in BC patients with BRCA1/2 mutation, the effectiveness of poly(ADP-ribose) polymerase (PARP) inhibitors contributes to the extension of disease-free survival. PARP1, an enzyme involved in DNA repair, participates in the correction of DNA single-strand breaks (SSBs), which, if unrepaired, may convert into double-stranded DNA breaks (DSBs) during DNA replication. DNA replication-derived DSBs result in chromosome breakages and translocations, leading to severe genome instability [10]. The pivotal mechanism in repairing DSBs is the homologous recombination (HR) leading to the replacement of DSBs with an undamaged sequence [11,12,13,14]. Inactivating mutations in HR genes leads to the accumulation of other mutations in the genome, causing changes that promote carcinogenesis. The therapy of HR-deficient BC patients with PARP1 inhibitors (PARPi) causing synthetic lethality results in deleterious accumulation of mutations and, thereafter, apoptosis [15, 16]. Thus, PARP inhibitors are effective in the therapy of patients with BRCA1/2mut breast cancer and homologous recombination deficiency (HRD) [5, 15, 16].
Many studies confirm that BRCA-dependent carcinogenesis is related to HR system inactivation [11, 17, 18]. Other HR genes involved in BC development include PALB2, BARD1, TP53, BRIP1, and RAD51C [19]. Previous research revealed that mutations in the above-mentioned genes significantly decrease DSB repair activity [20].
Our study aims to evaluate the alterations in the expression/transcript level of the following HR genes: BRCA1, BRCA2, ATM, BARD1, FANCA, FANCB, FANCI, RAD50, RAD51D, BRIP1, and CHEK2. As a result, we aim to develop a potential prognostic marker in patients diagnosed with BRCA-positive BC.
Material
Breast cancer tissue samples were secured by the core needle biopsy before the patient's systemic treatment to assess the expression level of selected HR genes in tumour cells.
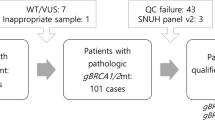
Core needle and vacuum-assisted breast biopsy (CNB and VABB) samples from 45 BC patients were collected and preserved in formalin-fixed paraffin-embedded (FFPE) blocks for subsequent RNA extraction and analysis. A pathologist selected the most representative BC tissue sections containing approximately 50% of cancer cells (macrodissection). All samples were taken before the chemotherapy. Later, all patients received treatment based on standard chemotherapy regimens (anthracyclines, taxanes).
In all patients, partial (PR) or complete pathological response (CR) was observed after neoadjuvant therapy (11 PR vs 33 CR). The complete pathological response was defined as the disappearance of all invasive cancer tissue in the resected breast specimen and in all sampled regional lymph nodes after the completion of neoadjuvant chemotherapy.
All patients were referred for a consultation with a clinical geneticist regarding hereditary breast and ovarian cancer predisposition. In the case of the germline BRCA1/2 pathogenic or likely pathogenic variant, consultation with a clinical geneticist and the appropriate molecular test were also offered to the patient’s adult close relatives.
Ethics statement
All patients signed informed consent. The study was approved by the Ethics Committee of Wroclaw Medical University (Nos. 611/2019 and 65/2023). All patients were diagnosed and treated in the Breast Unit, Lower Silesian Oncology, Pulmonology, and Hematology Center, Wroclaw, Poland. All samples were taken as part of the patient’s diagnostic and therapeutic scheme. All procedures performed in this study followed the principles for medical research of the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Methods
The pathogenic BRCA1 or BRCA2 germinal variants were assessed from blood using NGS BRCA1/2 panel testing (Devyser BRCA NGS CE-IVD, MiSeq Dx Illumina). Polish recurrent pathogenic variants were estimated/assessed using a laboratory-developed PCR screening test, whose results were confirmed by Sanger sequencing (BigDye™ Terminator v3.1 Cycle Sequencing Kit, ThermoFisher Scientific; Termocykler C1000 Touch Thermal Cycler, BioRad; ABI 3500 Series Genetic Analyzer, ThermoFisher Scientific).
RNA extraction
Total RNA was extracted from breast cancer FFPE tissue sections using a Qiagen® RNeasy FFPE Mini Kit according to the manufacturer’s instructions. The RNA quality was determined using a NanoPhotometer N60 (Implen). The samples with an RNA concentration of ≥ 60 ng/µl, an A260/A280 ratio of 1.8 ~ 2.0, and an A260/A230 ratio of 2 ~ 2.2 were accepted for the analysis.
PCR array
The Qiagen® Custom RT2 Profiler PCR Array was used to analyse the expression level of 11 HR genes (BRCA1, BRCA2, ATM, BARD1, FANCA, FANCB, FANCI, RAD50, RAD51D, BRIP1, and CHEK2) and two or three housekeeping genes (GAPDH and B2M, and for BRCA1/2 additionally ACTB). All genes were analysed in triplicates with respective housekeeping genes and controls included in the PCR plate according to the producer protocol using the real‐time cycler Bio-Rad CFX96 (Fig. 1).

Representations of real-time PCR amplification curves. Three replicates of the amplification reaction for each sample are shown either on a linear scale (left) or on a semi-log scale (right). Figure 1A The plot for BRCA1 mRNA expression in BRCA1-mutated tissue (Cq: 31.31/31.55/31.39); Fig. 1B The plot for BRCA1 mRNA expression in BRCA2-mutated tissue (Cq: 30.15/30.19/30.52); Fig. 1C The plot for BRCA1 mRNA expression in BRCA1/2-unmutated tissue (Cq: 36.65/34.89/35.82)
Statistical analysis
Data analysis was performed using R/Bioconductor environment and HTqPCR and compareGroups packages [PMID: 19,808,880; https://www.jstatsoft.org/article/view/v057i12]. Raw data were normalised using the B2M gene as a housekeeping reference. Data distribution of continuous variables was assessed using the Shapiro–Wilks test. Depending on whether the variable was considered as continuously normal-distributed, continuous non-normal-distributed, or categorical t-test, the Kruskal–Wallis test or chi-squared test were used, respectively. The significance level was set at 0.05.
Results
The study group consisted of 45 women aged 35–76 years (average age 53 years), including 24 women with a confirmed diagnosis of breast cancer and BRCA1/2 germline mutation and 21 women diagnosed with invasive breast cancer and no BRCA1/2 germline mutations.
All patients received systemic neoadjuvant therapy. The receptor status (ER, PR, and HER2), Ki67 status, adjuvant therapy response, and histopathological classification are presented in Table 1
No significant difference has been found for ATM, BARD1, FANCA, FANCB, FANCI, RAD50, RAD51D, BRIP1, and CHEK2 expression in BRCA1/2mut cancer tissues as compared to BRCA1/2wt samples (Table 2, Fig. 2).

Box plots representing expression levels of selected HR genes considering germline BRCA1/2 mutation status
The only statistically significant difference was observed for the BRCA1 gene expression. In BRCA1mut cancer tissues, the BRCA1 expression level was significantly higher than in BRCA2mut and BRCA1/2wt cancer tissues (Fig. 1). Apart from this observation, we found that BRCA1mut cancer tissues displayed significantly higher expression of Ki67 and a significantly higher proportion of ER and PR negative cases. Moreover, patients with BRCA1mut responded better to the applied therapy (higher proportion of complete response) than BRCA2mut and/or BRCA1/2wt patients (Table 3).
Discussion
Transcriptome profiling may be a powerful and effective method for searching for new molecular biomarkers of cancer prognosis, risk of progression, cancer-free survival, and other clinical features. Recent studies confirmed that a high homologous recombination deficiency (HRD) score is associated with poor survival among BC, prostate cancer, glioma, and head and neck squamous cell carcinoma (HNSCC) patients [21,22,23,24,25]. Hence, HRD, resulting from the loss of function of HR genes, including BRCA1/2, has been approved as an independent predictive biomarker of sensitivity to PARPi therapy [15, 16, 26]. Moreover, recent studies suggest a potential predictive value of HRD status in platinum-based chemotherapy in breast [27] and ovarian cancer (OC) patients [28].
Our study assessed the expression of the selected main HR pathway genes in cancer tissue to check whether their mRNA levels are altered in the BRCA1/2mut BC. The rationale for this research was based on the extensive interconnections among HR proteins, where deregulation of a single but critical gene’s expression could potentially impact the expression of others.
The only statistically significant differences were observed for BRCA1, as its mRNA level was elevated in BRCA1-mutated tissues compared to BRCA2-mutated and BRCA1/2 wild-type tissues.
Our results are consistent with those published by Wang et al., who proved that BRCA1 and BRCA2 gene expression is upregulated in breast and ovarian cancer (OC) tissues. Moreover, these authors observed an increased expression of NF1 and SYCP2 genes, interacting with BRCA1/2 genes in the regulation of the cell cycle. Therefore, they suggested the importance of functional interrelations among the BRCA1/2 with the other genes involved in BC and OC development and progression, thus, influencing the clinical course of disease and treatment outcomes [29].
In their recent study on 38 ovarian cancer vs 11 fallopian tube tissues, Custódio et al. showed that BRCA1/2 mRNA expression varied between individual samples. Moreover, in tissues characterised by downregulated BRCA1/2 expression, the other 12 genes involved in the HR pathway also exhibited low mRNA levels. The analysis of 299 ovarian cancer samples from The Cancer Genome Atlas (TCGA) confirmed these findings [30]. It is important to emphasise that our study’s findings on BRCAmut breast cancer tissues differ, as we observed elevated mRNA levels for BRCA1. This discrepancy may arise from the varying schemes and methodologies employed in the studies: 1) the cancer type (BC vs OC) or 2) different HR genes studied (ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERCC6, ERCC8, EXO1, FAN1, FANCA, FANCB, FANCC vs ATM, BARD1, FANCA, FANCB, FANCI, RAD50, RAD51D, BRIP1, CHEK2), 3) different BRCA1/2 mutational status (17/40 (42.5%) samples with BRCA1/2 pathogenic or likely pathogenic variant vs 24/45 (53%)), and finally, the most importantly 4) different laboratory methods (very sensitive and accurate NanoString Technology and droplet digital PCR (ddPCR) vs real-time PCR).
Another study on 96 fresh frozen ovarian cancer tissues obtained from chemotherapy-naïve patients and patients after neoadjuvant chemotherapy was performed using a tailored NanoString-based Pancancer Pathway Panel of 19 HR genes and showed a correlation between over-expression of C11osf30, NBN, FANCF, FANCC, FANCB, RAD50 and improved outcome in chemotherapy-naïve patients. Moreover, a correlation has been observed between over-expression of BRCA2, TP53, FANCB, and RAD51 and worse outcomes in chemotherapy-treated patients. When adding the extent of debulking as a covariate, the expression of NBN, FANCF, RAD50, and RAD51 was significant, respectively [31].
Also, in the bladder cancer cell lines, the expression of four DNA damage repair genes, including two MR genes, was evaluated respectively before and after chemotherapy. The authors of this study revealed that the increase of BRCA1 and RBBP8 expression induced by chemotherapy correlates with worse sensitivity to treatment in non-basal and non-luminal cell lines. In contrast, no significant differences in the basal-cell lines most sensitive to chemo and radiotherapy were observed. This observation revealed the high diversity of HR genes expression, which correlates with the histopathological characteristics of tumours [32]. Moreover, in 413 bladder cancer samples (data derived from TCGA), a significantly higher expression level of four genes, RAD21, RAD51, BARD1, and ERBB2, was observed in ERBB-low as compared to ERBB-high tumours. Also, the combined expression of two out of four tested genes has been shown to correlate with chosen clinical features. This confirms the interconnections among the expression of different HR pathway genes [33].
In sporadic gastric cancer patients after receiving postoperative adjuvant chemotherapy, BRCA1/BRCA2 expression assessed using IHC and mRNA tests displayed a correlation between BRCA2-elevated expression with advanced tumour stage but not disease-free and overall survival [34].
These findings indicate that the investigation should encompass not only the alterations in individual HR genes but also their interrelations, considering the clinical course of the disease alongside histopathological and biochemical variables characterising the studied cancer.
Conclusion
The mRNA expression of BRCA1 is upregulated in breast cancer tissues harbouring BRCA1 mutation. However, no relevant differences in the expression of other HR genes have been observed between BRCA1/BRCA2mut and non-mutated BC tissues. Hence, it appears that BRCAmut tissues do not exhibit crucial compensatory alterations in the mRNA expression of other HR genes.
Limitations
The prognostic value of HR gene expression in BC could not be assessed due to the limited sample size and the variability in histopathological classifications.
Data availability
No datasets were generated or analysed during the current study.
References
Nardin S, Mora E, Varughese FM et al (2020) Breast cancer survivorship, quality of life, and late toxicities. Front Oncol. https://doi.org/10.3389/fonc.2020.00864
Wasserman, A., Wasserman, E., & Mak, R. (2022) Recent Advances In Breast Cancer Treatments. Journal of Student Research, 10(4). https://doi.org/10.47611/jsrhs.v10i4.2123
Miglietta F, Bottosso M, Griguolo G et al (2022) Major advancements in metastatic breast cancer treatment: when expanding options means prolonging survival. ESMO Open. 7:100409
Yersal O, Barutca S (2014) Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol 5:412. https://doi.org/10.5306/WJCO.V5.I3.412
Carvalho FM (2023) Triple-negative breast cancer: from none to multiple therapeutic targets in two decades. Front Oncol. https://doi.org/10.3389/fonc.2023.1244781
Zhao Y, Xiong D, Yang B et al (2023) Application of multigene panel detection in breast cancer. J Pak Med Assoc 73:1862–1868
Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair and mutagenesis. Environ Mol Mutagen 58:235. https://doi.org/10.1002/EM.22087
Wang R, Sun Y, Li C et al (2023) Targeting the DNA damage response for cancer therapy. Int J Mol Sci 24:15907
Huang R, Zhou PK (2021) DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther. https://doi.org/10.1038/s41392-021-00648-7
Ensminger M, Iloff L, Ebel C et al (2014) DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J Cell Biol 206:29. https://doi.org/10.1083/JCB.201312078
Creeden JF, Nanavaty NS, Einloth KR et al (2021) Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer. https://doi.org/10.1186/S12885-021-08863-9
Doig KD, Fellowes AP, Fox SB (2023) Homologous recombination repair deficiency: an overview for pathologists. Mod Pathol 36:100049
Wang Y, Ung MH, Cantor S, Cheng C (2017) Computational investigation of homologous recombination DNA repair deficiency in sporadic breast cancer. Sci Rep. https://doi.org/10.1038/s41598-017-16138-2
Yamamoto H, Hirasawa A (2021) Homologous recombination deficiencies and hereditary tumors. Int J Mol Sci 23:348. https://doi.org/10.3390/IJMS23010348
Chopra N, Tovey H, Pearson A et al (2020) Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun. https://doi.org/10.1038/s41467-020-16142-7
Dilmac S, Ozpolat B (2023) Mechanisms of PARP-inhibitor-resistance in BRCA-mutated breast cancer and new therapeutic approaches. Cancers (Basel) 15:3642
Royfman R, Whiteley E, Noe O et al (2021) BRCA1/2 signaling and homologous recombination deficiency in breast and ovarian cancer. Future Oncol 17:2817–2830. https://doi.org/10.2217/FON-2021-0072
Ladan MM, van Gent DC, Jager A (2021) Homologous recombination deficiency testing for BRCA-like tumors: the road to clinical validation. Cancers (Basel) 13:1–23. https://doi.org/10.3390/CANCERS13051004
Sato K, Koyasu M, Nomura S et al (2017) Mutation status of RAD51C, PALB2 and BRIP1 in 100 Japanese familial breast cancer cases without BRCA1 and BRCA2 mutations. Cancer Sci 108:2287. https://doi.org/10.1111/CAS.13350
Jachimowicz RD, Goergens J, Reinhardt HC (2019) DNA double-strand break repair pathway choice - from basic biology to clinical exploitation. Cell Cycle 18:1423–1434
Moutafi M, Economopoulou P, Rimm D, Psyrri A (2021) PARP inhibitors in head and neck cancer: molecular mechanisms, preclinical and clinical data. Oral Oncol 117:105292
Sim HW, Galanis E, Khasraw M (2022) PARP inhibitors in glioma: a review of therapeutic opportunities. Cancers (Basel) 14:1003
Zhu S, Zhao J, Nie L et al (2022) Homologous recombination deficiency (HRD) score in aggressive prostatic adenocarcinoma with or without intraductal carcinoma of the prostate (IDC-P). BMC Med. https://doi.org/10.1186/s12916-022-02430-0
Toh MR, Ngeow J (2021) Homologous recombination deficiency: cancer predispositions and treatment implications. Oncologist 26:e1526–e1537. https://doi.org/10.1002/onco.13829
Singh DD, Parveen A, Yadav DK (2021) Role of parp in tnbc: Mechanism of inhibition, clinical applications, and resistance. Biomedicines. 9:1512
Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E (2020) PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol. 8:564601. https://doi.org/10.3389/fcell.2020.564601
Galland L, Roussot N, Desmoulins I et al (2023) Clinical utility of genomic tests evaluating homologous recombination repair deficiency (HRD) for treatment decisions in early and metastatic breast cancer. Cancers (Basel) 15:1299
Kekeeva T, Andreeva Y, Tanas A et al (2023) HRD testing of ovarian cancer in routine practice: what are we dealing with? Int J Mol Sci. https://doi.org/10.3390/ijms241310497
Wang Z, Zhang J, Zhang Y et al (2018) Expression and mutations of BRCA in breast cancer and ovarian cancer: evidence from bioinformatics analyses. Int J Mol Med 42:3542–3550. https://doi.org/10.3892/ijmm.2018.3870
Custódio N, Savisaar R, Carvalho C et al (2022) Expression profiling in ovarian cancer reveals coordinated regulation of BRCA1/2 and homologous recombination genes. Biomedicines. https://doi.org/10.3390/biomedicines10020199
Kessous R, Octeau D, Klein K et al (2018) Distinct homologous recombination gene expression profiles after neoadjuvant chemotherapy associated with clinical outcome in patients with ovarian cancer. Gynecol Oncol 148:553–558. https://doi.org/10.1016/j.ygyno.2018.01.017
Sun S, Jiang K, Zeng J (2022) Differential expression of DNA damage repair genes after chemoradiotherapy and inhibition rate in different bladder cancer cells. Transl Androl Urol 11:1336–1344. https://doi.org/10.21037/tau-22-543
Albarakati N, Al-Ghamdi H, Al-Sowayan B, Alshareeda A (2023) Homologous recombination mRNAs (RAD21, RAD50 and BARD1) have a potentially poor prognostic role in ERBB2-low bladder cancer patients. Sci Rep. https://doi.org/10.1038/s41598-023-38923-y
Kim HS, Hwang IG, Min HY et al (2019) Clinical significance of BRCA1 and BRCA2 mRNA and protein expression in patients with sporadic gastric cancer. Oncol Lett 17:4383–4392. https://doi.org/10.3892/ol.2019.10132
Funding
This research was financed through statutory subsidies by the Minister of Health as part of the research grants SUBK.A290.22.077, SUBZ.A290.23.067, and SUBZ.C280.24.063 (record number in the Simple System).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Rafal Matkowski, Piotr Kasprzak, Bartlomiej Szynglarewicz, Ewelina Iwaneczko, and Stanislaw Supplitt performed material preparation and clinical data collection. Mariola Abrahamowska, Ewelina Czykalko, and Izabela Laczmanska executed the laboratory experiments. Lukasz Laczmanski and Pawel Karpinski performed data analysis. Izabela Laczmanska and Stanislaw Supplitt wrote the first draft of the manuscript. Izabela Laczmanska corrected the manuscript after revision. Maria M. Sasiadek and Adam Maciejczyk revised the manuscript critically for important intellectual content. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Izabela, L., Rafal, M., Stanislaw, S. et al. Alterations in the expression of homologous recombination repair (HRR) genes in breast cancer tissues considering germline BRCA1/2 mutation status. Breast Cancer Res Treat (2024). https://doi.org/10.1007/s10549-024-07441-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10549-024-07441-4