
Abstract
Purpose
CompLEEment-1 is a phase 3b trial in an expanded patient population with hormone receptor-positive (HR +), human epidermal growth factor receptor-2–negative (HER2–) advanced breast cancer (ABC), the largest current trial of cyclin-dependent kinase 4 and 6 inhibitors in ABC.
Methods
Patients treated with ≤ 1 line of prior chemotherapy and no prior endocrine therapy for ABC received ribociclib 600 mg/day (3-weeks-on/1-week-off) plus letrozole 2.5 mg/day and additionally monthly goserelin/leuprolide in men and pre-/perimenopausal women. Eligibility criteria allowed inclusion of patients with stable CNS metastases and an Eastern Cooperative Oncology Group performance status of 2. Primary objectives were safety and tolerability, and secondary objectives were efficacy and quality of life (QoL).
Results
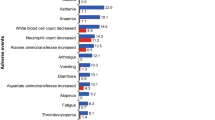
Overall, 3,246 patients were evaluated (median follow-up 25.4 months). Rates of all-grade and grade ≥ 3 treatment-related adverse events (AEs) were 95.2% and 67.5%, respectively. Treatment-related discontinuations due to all grade and grade ≥ 3 AEs occurred in 12.9% and 7.3% of patients, respectively. Rates of all-grade AEs of special interest (AESI) were as follows: neutropenia (74.5%), increased alanine aminotransferase (16.2%), increased aspartate aminotransferase (14.1%), and QTcF prolongation (6.7%); corresponding values for grade ≥ 3 AESI were 57.2%, 7.7%, 5.7%, and 1.0%, respectively. Median time to progression was 27.1 months (95% confidence interval, 25.7 to not reached). Patient QoL was maintained during treatment.
Conclusion
Safety and efficacy data in this expanded population were consistent with the MONALEESA-2 and MONALEESA-7 trials and support the use of ribociclib plus letrozole in the first-line setting for patients with HR + , HER2– ABC.
Trial registration
linicalTrials.gov NCT02941926.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Approximately 75% of advanced breast cancers (ABCs) are hormone receptor‑positive (HR +) [1,2,3]. For many years, endocrine therapy (ET) has been the treatment of choice for patients with HR + ABC; however, resistance remains a barrier to long-term clinical benefit, leading to the development of therapies that reverse or delay this resistance [4]. The combination of ET with cyclin-dependent kinases 4/6 inhibitors (CDK4/6is) is the recommended first-line treatment for patients with HR + , human epidermal growth factor receptor-2–negative (HER2–) ABC, in the absence of visceral crisis [5, 6].
In multiple phase 3 clinical trials in HR + , HER2– ABC, ribociclib (a selective, small-molecule CDK4/6i) in combination with ET has demonstrated superior clinical benefit compared with endocrine monotherapy, including significantly better overall survival (OS) in premenopausal women when given with a non-steroidal aromatase inhibitor (NSAI) (MONALEESA-7) [7] and in postmenopausal women in combination with fulvestrant (MONALEESA-3) [8]. The ongoing phase 3 MONALEESA-2 study is investigating ribociclib with letrozole vs letrozole alone in postmenopausal women. Although OS results are awaited, MONALEESA-2 has reported a significant improvement in progression-free survival (PFS) in the ribociclib arm (25.3 months vs 16.0 months, hazard ratio = 0.568; 95% confidence interval [CI], 0.457–0.704; P = 9.63 × 10–8) [9]. Clinical trials, however, are often limited to patients with good performance status and frequently exclude patients with central nervous system (CNS) metastases and patients who have received chemotherapy for ABC [10, 11]. Furthermore, some patient populations (e.g., men with breast cancer) are small and their representation in ABC trials is uncommon [12].
Data from clinical practice indicate that a significant proportion of patients with HR + , HER2– ABC still receive chemotherapy as first-line treatment [13,14,15,16], while international guidelines recommend this only in the presence of visceral crisis [5]. Notably, premenopausal women and those with visceral or CNS metastases are understood to have worse outcomes when treated with ET and may be more likely to receive chemotherapy as first-line treatment for HR + , HER2– ABC [16]. Further information is needed to better understand the safety and efficacy of ET plus CDK4/6is in these patients.
The CompLEEment-1 trial investigated the safety and efficacy of ribociclib in combination with letrozole in a large, diverse patient cohort, representative of real‑world clinical practice, who had not received prior ET for advanced disease. The trial included a Core Phase (time from first patient/first visit to 18 months after the last patient/first visit) and a follow-on Extension Phase to last patient/last visit. Here, we report safety and efficacy results from the Core Phase.
Methods
Study design and treatment
CompLEEment-1 is a multicenter phase 3b trial (see Online Resource 1) designed to evaluate the overall safety, tolerability, and clinical efficacy of ribociclib in combination with letrozole in pre-/postmenopausal women and men with HR + , HER2− ABC and no prior ET for advanced disease. Patients were treated with ribociclib (starting dose: 600 mg orally, once daily, 3-weeks-on/1-week-off) plus letrozole (2.5 mg orally, once daily, on a continuous schedule) with or without food. Pre-/perimenopausal women and men also received goserelin (3.6 mg subcutaneously) or leuprolide (7.5 mg intramuscularly; added per protocol amendment in response to guideline updates [6, 17], administered on Day 1 of Cycle 1 and every 28 days thereafter). Treatment continued until disease progression, unacceptable toxicity, death, or discontinuation from study treatment for any other reason. Patients who discontinued ribociclib were discontinued from the study and censored. All patients were followed for 30 days following the last ribociclib dose. The Supplement includes further information on criteria for dose interruption, reduction, or permanent discontinuation due to adverse events (AEs) (see Online Resources 8–11), medications prohibited during this study (See Online Resource 12), and patient selection criteria.
Endpoints
The primary endpoint was safety/tolerability, measured by the number of patients who experienced: any AEs; grade 3/4 AEs; serious AEs (SAEs); AEs of special interest (AESI); AEs leading to dose reduction, interruption, or discontinuation; and AE-related deaths. AESIs were defined according to ongoing reviews of all ribociclib safety data, including neutropenia, QT interval corrected for heart rate using Fridericia’s formula (QTcF) prolongation, and hepatobiliary toxicity. In an exploratory analysis, exposure-adjusted AEs were also evaluated. Acceptable safety/tolerability constituted similar results to those observed in the MONALEESA trials.
Secondary endpoints related to efficacy were time to progression (TTP) based on investigator assessment, overall response rate (ORR) for patients with measurable disease, and clinical benefit rate (CBR); PFS, although not included as a predefined endpoint, was evaluated. Efficacy and response classifications are defined in Online Resource 2. Other secondary endpoints included patient-reported outcome (PRO) measures of health-related quality of life (HRQoL) using the Functional Assessment of Cancer Therapy–Breast Cancer (FACT-B) questionnaire; due to the nature of the questionnaire and validation, it was only completed by female patients. Data were collected electronically in pre-selected countries: the USA, Canada, the UK, France, Italy, and Spain. FACT-B data were entered by patients at the clinic.
Assessments
Safety was monitored by assessing patient symptoms through physical exams, including measurement of vital signs, cardiac assessment (i.e., 12-lead electrocardiogram), and assessing biochemical and hematologic laboratory values at various timepoints during the Core Phase; details of the assessment schedule are shown in Online Resource 2. AEs were characterized and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), v4.03 [18].
Tumor response was assessed locally, based on Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Tumor assessments were performed according to the current standard of care; assessments are recommended to take place every 12 weeks until disease progression (See Online Resource 2). There was no planned central review of imaging assessments.
Statistical analysis
The safety analysis and full analysis sets were used for statistical analysis and data reporting. The safety analysis (safety outcomes) and full analysis (efficacy outcomes) sets comprised patients who received ≥ 1 dose of either ribociclib or letrozole or goserelin/leuprolide (if applicable) in the Core Phase.
The primary endpoint of safety/tolerability (number [%] of AEs, grade 3/4 AEs, and SAEs, AESI and AEs leading to treatment discontinuation and deaths, and AEs leading to dose reduction or dose interruption) was summarized descriptively in the safety analysis set. Attempts were made to ensure that comprehensive information was obtained for safety data and no imputation was applied for missing data. Data obtained for relative dose intensity (RDI) considered both zero and non-zero dose days and therefore accounted for dose interruptions.
For the secondary endpoint of TTP and for the post hoc, exploratory analysis of PFS, distribution was estimated using the Kaplan–Meier method and medians were presented with 95% CIs [19]. The other secondary endpoints of ORR and CBR were summarized using descriptive statistics (N [%]) combined with 2-sided exact binomial 95% CIs [20].
The PRO analysis set comprised female patients in pre-selected countries for whom baseline and ≥ 1 post-baseline PRO measurements were available. Descriptive statistics were used to summarize the FACT-B questionnaire subscale and overall scores at each scheduled assessment; the change from baseline at each assessment was summarized. Scores for a given scale/subscale were considered missing if > 50% of the items were missing; otherwise, the average of the non-missing items was used to impute missing items.
Results
Study population and disposition
Overall, 3,246 patients were enrolled and received ≥ 1 dose of study treatment between November 30, 2016 and March 22, 2018. The cut-off date for this analysis was November 8, 2019, and the median duration of follow-up was 25.4 months (minimum 19.1 months).
At data cutoff, 1,301 (40.1%) patients had completed Core Phase treatment, 415 of whom moved to the Extension Phase. Overall, 1,945 (59.9%) patients permanently discontinued treatment, mostly due to progressive disease (34.2%) and AEs (15.5%).
Patient age ranged from 20 to 92 years (median, 58.0 years). Patient populations of special interest, including patients ≥ 75 years of age, premenopausal female patients, male patients, patients with de novo ABC, patients with CNS metastases, patients with an Eastern Cooperative Oncology Group (ECOG) performance status 2, and patients who had received prior chemotherapy for ABC, are described in Table 1. At baseline, 2079 (64.0%) patients had measurable disease.
The median duration of exposure to ribociclib was 17.5 months and the median and mean RDIs were 95.2% and 86.4%, respectively. The median duration of exposure to letrozole was 17.7 months. The average daily dose of ribociclib was 547.7 mg (median 600.0 mg).
Primary endpoint
At the data cut-off date, 3,203 (98.7%) patients had experienced an AE and 2,461 (75.8%) patients had experienced a grade ≥ 3 AE. The most common (≥ 20%) all‑grade AEs (regardless of causality) were neutropenia (n = 2,417; 74.5%), nausea (n = 1166; 35.9%), leukopenia (n = 887; 27.3%), fatigue (n = 760; 23.4%), diarrhea (n = 690; 21.3%), arthralgia (n = 677; 20.9%), and vomiting (n = 649; 20.0%), whereas the most common (≥ 5%) all-cause grade ≥ 3 AEs were neutropenia (n = 1856; 57.2%), leukopenia (n = 345; 10.6%), alanine aminotransferase (ALT) increased (n = 249; 7.7%), and aspartate aminotransferase (AST) increased (n = 184; 5.7%) (Table 2). Thirty-six (1.1%) patients experienced febrile neutropenia. The number of patients who discontinued due to an AE (regardless of causality) was 528 (16.3%), with the most common AEs (regardless of causality) causing discontinuation being ALT increased (197; 6.1%), AST increased (129; 4.0%), and transaminases increased (22; 0.7%).
In total, 3091 (95.2%) patients experienced a treatment-related AE, 2192 (67.5%) patients experienced a grade ≥ 3 treatment-related AE, and 203 (6.3%) patients experienced a treatment-related SAE (Table 2). Treatment-related all grade and grade ≥ 3 AEs led to dose adjustment/interruption for 2,235 (68.9%) and 1,964 (60.5%) patients, respectively. In total, 418 (12.9%) patients had treatment-related AEs (regardless of causality) leading to discontinuation, and 237 (7.3%) patients had grade ≥ 3 treatment-related AEs leading to discontinuation. Treatment-related grade 5 SAEs are presented in Online Resource 13.
Values for all grade and grade ≥ 3 AESIs, respectively, were as follows: 217 patients (6.7%) and 33 patients (1.0%) experienced QTcF prolongation, 526 patients (16.2%) and 249 patients (7.7%) experienced increased ALT, and 459 patients (14.1%) and 184 patients (5.7%) experienced increased AST (neutropenia values have already been stated) (See Online Resource 14). A > 60 ms increase from baseline in QTcF interval was observed in 189 (5.9%) patients, whereas a new QTcF of > 480 to ≤ 500 ms and > 500 ms was observed in 59 (1.8%) and 42 (1.3%) patients, respectively.
There were 1,716 (52.9%) patients and 597 (18.4%) patients who, because of neutropenia, required a ribociclib dose interruption or reduction, respectively. Otherwise, dose interruptions or reductions due to AESIs were rare. The numbers of patients who experienced permanent drug discontinuation because of neutropenia, ALT increased, AST increased, and QTcF prolongation were 18 (0.6%), 197 (6.1%), 129 (4.0%), and 8 (0.2%), respectively. At the data cutoff, the rate of recovery/resolution was greater than the rate of non-recovery/non-resolution for all AESIs (neutropenia, 70.1% vs 39.9%; ALT increased, 10.6% vs 8.0%; AST increased, 9.4% vs 7.1%; QTcF prolongation, 5.9% vs 1.0%). AESIs rarely led to hospitalization (0%–0.3%); no AESIs were fatal (Table 3). AESI events such as neutropenia, increased ALT, and increased AST, when adjusted for ribociclib exposure for time periods of 0–1 years, 1–2 years, and > 2 years, decreased in occurrence (Table 3). The median time to first occurrence of grade ≥ 3 neutropenia was 17.1 weeks (95% CI, 14.0–24.1). However, time to onset of grade ≥ 3 neutropenia decreased sharply after Week 8, and 60% event probability was not reached until after Week 80, while 70% event probability was not reached until after Week 128 (see Online Resource 3).
A total of 74 (2.3%) on-treatment deaths occurred up to the cut-off date (i.e., from the date of first administration of study treatment to 30 days after last administration of study treatment). ABC was the primary cause of death for 38 (1.2%) patients. Overall, 14 (0.4%) patients had a treatment-related fatal SAE; 22 deaths were unrelated to treatment.
Secondary and exploratory endpoints
Over a median follow-up of 25.4 months, the median TTP was 27.1 months (95% CI, 25.7 to not reached), whereas the estimated event-free probability at 24 months was 54.7% (95% CI, 52.5–56.8; Fig. 1). In total 2140 (65.9%) of patients were censored from the TTP analysis; the main reasons for censoring were ‘adequate assessment no longer available’ in 720 patients and ‘withdrawal of consent’ in 73 patients.

Kaplan–Meier plot of time to progression per local investigator assessment (full analysis set). CI confidence interval, NR not reached
ORRs for the total population and patients with measurable disease at baseline (n = 2079 [64.0%]) were 29.3% (95% CI, 27.7–30.9) and 43.6% (95% CI, 41.5–45.8), respectively. The CBRs were similar for the total study population and patients with measurable disease at baseline (70.7% [95% CI, 69.1–72.2] and 69.1% [95% CI, 67.1–71.1], respectively) (Table 4). In an exploratory analysis, median PFS was 26.7 months (95% CI, 24.8–30.1) for the total study population (See Online Resource 4).
The PRO analysis set included 1,230 patients from 6 pre-selected countries. Descriptive analysis of median delay to first occurrence of a clinically relevant deterioration from baseline (≥ 7-point decrease) [21] in overall FACT-B score was not reached (See Online Resource 5). Overall changes in FACT-B scores from baseline until end of treatment were assessed, as well as FACT-B scores for individual domains, including physical, social/family, emotional and functional well-being, and additional concerns (See Online Resource 15). Scores for emotional and functional well-being did not decrease below baseline levels while on treatment (See Online Resources 6 and 7).
Discussion
In the CompLEEment-1 study, ribociclib in combination with letrozole demonstrated safety and efficacy consistent with that seen in the pivotal phase 3 studies (MONALEESA-2 [9, 22] and MONALEESA-7 [7, 23]), in a much larger, diverse cohort of patients with HR + , HER2– ABC who had not previously received ET for advanced disease. The AE profile was manageable and no new safety signals were identified; in addition, median TTP was 27.1 months after a median follow-up of 25.4 months and patient HRQoL was not adversely impacted. The median PFS of 26.7 months is similar to that reported in the ribociclib arms of MONALEESA-2 (25.3 months [95% CI, 23.0–30.3]) [9] and MONALEESA-7 (23.8 months [95% CI, 19.2‑NR] [23]), indicating that the efficacy reported in phase 3 trials is achievable in real‑world practice.
The patient characteristics in CompLEEment-1 were more broadly representative of patients in clinical practice than those included in phase 3 trials of CDK4/6i combined with NSAI [7, 9, 24, 25]. The expanded patient population in CompLEEment-1 included patients treated with prior chemotherapy for advanced disease, patients with ECOG performance status 2, patients with stable CNS metastases, premenopausal women, and men—a larger population more representative of real‑world clinical practice, and one not well studied in randomized controlled trials with palbociclib and abemaciclib [22]. For example, 10.0% (324 patients) and 3.5% (112 patients) of those enrolled had received prior chemotherapy for advanced disease or had ECOG performance status 2, respectively. This represents a large group of patients and few trials have reported results in similarly broad populations. The study cohort was also diverse in terms of race and age. Compared with ribociclib-treated patients in MONALEESA-2, CompLEEment-1 included more patients with both disease-free interval ≤ 12 months (7.3% vs 1.2%) and ≥ 3 metastatic sites (43.3% vs 34.1%).
Many of the expected adverse reactions observed with ribociclib, including neutropenia, QTcF prolongation, and hepatobiliary adverse reactions, can be managed by following dose interruption or reduction guidelines as per the label and/or with medication [17, 26]. Although incidences of dose interruption and reduction due to neutropenia occurred in 52.9% and 18.4% of patients, respectively, incidences leading to drug withdrawal (0.6%) and additional medication (3.5%) were rare. Discontinuations because of AESIs were infrequent, and AESIs were resolved in most cases; hospitalizations resulting from these events were rare (< 1%). Furthermore, exposure-adjusted AE data suggest that occurrences of most AESIs decreased during ribociclib treatment.
Efficacy results relating to progression (TTP and PFS) in this study are conservative compared with those observed in MONALEESA-2, which included PFS events occurring after patients had discontinued ribociclib treatment (during post-treatment efficacy follow-up); these patients were censored in CompLEEment-1. Importantly, TTP only documents progression or death due to underlying cancer, whereas PFS documents progression or death due to any cause which precludes direct comparisons between these endpoints. The ORRs of the total population (29.3%) and patients with measurable disease (43.6%) are lower than those seen in ribociclib‑treated patients in MONALEESA-2 (42.5% in the total population, 54.5% in patients with measurable disease) and MONALEESA-7 (41% in the total population, 51% in patients with measurable disease) [7, 9, 23]. This may be due to certain between‑trial differences in baseline characteristics (e.g., prior chemotherapy exposure or more metastatic sites) that were less favorable in CompLEEment-1 vs the pivotal studies. However, when compared with the MONALEESA trials, lower ORRs did not appear to translate to shorter PFS in CompLEEment-1. A limitation of CompLEEment-1 is that response assessment timings allowed for different intervals according to the local standard of care. As with all prospective real-world studies, lack of randomization and a control arm necessitate the cautious interpretation of efficacy results.
Treatment guidelines recommend that, in addition to efficacy and safety, HRQoL is evaluated using validated PRO measures [5], e.g., the FACT-B questionnaire [27], which is frequently used in ABC trials. Baseline FACT-B domain scores were similar to those of other CDK4/6i trials in ABC [28]. The median delay to first occurrence of a clinically relevant deterioration (≥ 7-point decrease) in overall FACT-B score was not reached, implying that quality of life (QoL) was maintained while on treatment. Individual FACT-B domain scores for emotional and functional well-being did not appear to be impacted by treatment initiation and were maintained while on treatment, relative to baseline. Although QoL assessments must be interpreted cautiously in single-arm studies, overall FACT-B results suggest that patients’ HRQoL was maintained while taking ribociclib. This is consistent with QoL outcomes in MONALEESA-2 and complements the results observed in MONALEESA-7, the only CDK4/6i study with improved QoL in premenopausal patients [29, 30].
Conclusion
Results from CompLEEment-1, the largest prospective dataset on ribociclib and any other CDK4/6i (to our knowledge), and one of the largest trials ever published in ABC, support the manageable safety profile and efficacy of ribociclib in combination with letrozole as first-line treatment for HR + , HER2– ABC. In addition to providing insights into the clinical management and mitigation of common AEs with ribociclib, CompLEEment-1 reports a vital assessment of a diverse patient population in conditions simulating a real-world setting. These results build on the available knowledge from the extensive MONALEESA program, and would potentially support clinical decisions in daily practice. Outcomes in patient subgroups of special interest will be presented in due course.
Data availability
The data that support the findings of this study are available from Novartis Pharmaceuticals Corporation but restrictions apply to the availability of these data (https://www.novartisclinicaltrials.com/TrialConnectWeb/home.nov). Data are however available from the authors upon reasonable request and with permission of Novartis Pharmaceuticals Corporation.
Code availability
Not applicable.
Change history
08 October 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10549-021-06374-6
References
Anderson WF, Chatterjee N, Ershler WB, Brawley OW (2002) Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Breast Cancer Res Treat 76:27–36
Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62:233–247
Setiawan VW, Monroe KR, Wilkens LR et al (2009) Breast cancer risk factors defined by estrogen and progesterone receptor status: The multiethnic cohort study. Am J Epidemiol 169:1251–1259
Garcia-Becerra R, Santos N, Diaz L, Camacho J (2012) Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci 14:108–145
Cardoso F, Senkus E, Costa A et al (2018) 4th ESO–ESMO international consensus guidelines for advanced breast cancer (ABC 4). Ann Oncol 29:1634–1657
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Breast Cancer, version 1.2020.
Im SA, Lu YS, Bardia A et al (2019) Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med 381:307–316
Slamon DJ, Neven P, Chia S et al (2019) Overall survival (OS) results of the phase III MONALEESA-3 trial of postmenopausal patients (pts) with hormone receptor-positive (HR+), human epidermal growth factor 2-negative (HER2−) advanced breast cancer (ABC) treated with fulvestrant (FUL) ± ribociclib (RIB). Ann Oncol 30:v851–v934
Hortobagyi GN, Stemmer SM, Burris HA et al (2018) Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol 29:1541–1547
Aapro MS, Kohne CH, Cohen HJ, Extermann M (2005) Never too old? Age should not be a barrier to enrollment in cancer clinical trials. Oncologist 10:198–204
Lin NU, Amiri-Kordestani L, Palmieri D, Liewehr DJ, Steeg PS (2013) CNS metastases in breast cancer: old challenge, new frontiers. Clin Cancer Res 19:6404–6418
Moelans CB, de Ligt J, van der Groep P et al (2019) The molecular genetic make-up of male breast cancer. Endocr Relat Cancer 26:779–794
Brufsky AM (2015) Delaying chemotherapy in the treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Med Insights Oncol 9:137–147
Burton T, Byfield SD, Smith GL et al (2016) Clinical and economic outcomes by first-line treatment among women with HR+/HER2- metastatic breast cancer in a large US health plan database. Curr Med Res Opin 32:1417–1423
Mitra D, Kurosky S, Zanotti G, Kaye J (2016) Real-world treatment patterns and clinical outcomes in ER+/HER2- metastatic breast cancer: Results from a multicountry retrospective medical record review. Value Health. https://doi.org/10.1016/j.jval.2016.03.1644
Jacquet E, Lardy-Cleaud A, Pistilli B et al (2018) Endocrine therapy or chemotherapy as first-line therapy in hormone receptor-positive HER2-negative metastatic breast cancer patients. Eur J Cancer 95:93–101
Novartis Europharm Limited. KISQALI (ribociclib) summary of product characteristics. https://www.ema.europa.eu/documents/product-information/kisqali-epar-product-information_en.pdf Accessed February 5, 2019.
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–47
Brookmeyer R, Crowley J (1982) A confidence interval for the median survival time. Biometrics 38:29–41
Clopper CJ, Pearson ES (1934) The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 26:404–413
Eton DT, Cella D, Yost KJ et al (2004) A combination of distribution- and anchor-based approaches determined minimally important differences (MIDs) for four endpoints in a breast cancer scale. J Clin Epidemiol 57:898–910
Hortobagyi GN, Stemmer SM, Burris HA et al (2016) Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med 375:1738–1748
Tripathy D, Im SA, Colleoni M et al (2018) Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol 19:904–915
Rugo HS, Finn RS, Dieras V et al (2019) Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat 174:719–729
Goetz MP, Toi M, Campone M et al (2017) MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 35:3638–3646
Novartis Pharmaceuticals Corporation. KISQALI (ribociclib) prescribing information. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. Accessed February 5, 2019.
Brady MJ, Cella DF, Mo F et al (1997) Reliability and validity of the functional assessment of cancer therapy-breast quality-of-life instrument. J Clin Oncol 15:974–986
Rugo HS, Dieras V, Gelmon KA et al (2018) Impact of palbociclib plus letrozole on patient-reported health-related quality of life: results from the PALOMA-2 trial. Ann Oncol 29:888–894
Verma S, O’Shaughnessy J, Burris HA et al (2018) Health-related quality of life of postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer treated with ribociclib + letrozole: Results from MONALEESA-2. Breast Cancer Res Treat 170:535–545
Harbeck N, Franke F, Villanueva-Vazquez R et al (2020) Health-related quality of life in premenopausal women with hormone-receptor-positive, HER2-negative advanced breast cancer treated with ribociclib plus endocrine therapy: Results from a phase III randomized clinical trial (MONALEESA-7). Ther Adv Med Oncol 12:1758835920943065
Acknowledgements
The authors would like to thank the patients enrolled in this study and their families. Medical editorial assistance was provided by Joe Hodgson BVM BVS of Healthcare Consultancy Group LLC.
Funding
Supported by Novartis Pharmaceuticals, which also funded medical writing assistance.
Author information
Authors and Affiliations
Contributions
Conception and design: CZ. Provision of study material or patients: EW, WJ, De MDL, PC, CZ, JSB, SD, MM, and SB. Acquisition, analysis, or interpretation of data: EW, JSB, and SB. Data analysis and interpretation: WJ, HAA, JL, MDL, PC, AR, JW, CZ, SD, MM, KZ, and LM-S. Drafting of the manuscript: EW, WJ, HAA, JL, MDL, PC, AR, CZ, SC, JSB, SD, and MM. Critical revision of the manuscript for important intellectual content: EW, WJ, HAA, JL, MDL, PC, AR, CZ, SC, JSB, SD, MM, MC, SB, EC, Wu, Zhou, and LM-S. Accountable for all aspects of the work: WJ, HAA, JL, MDL, PC, CZ, and JSB.
Corresponding author
Ethics declarations
Conflict of interest
Michelino De Laurentiis has received honoraria for consulting or advisory roles from Amgen, AstraZeneca, Celgene, Eisai, Lilly, MSD, Novartis, Pfizer, Pierre Fabre, and Roche; Honoraria: Amgen, AstraZeneca, Celgene, Eisai, Lilly, MSD, Novartis, Pfizer, Pierre Fabre, and Roche; honoraria for participation in speakers’ bureau from Novartis; research funding from Bristol-Myers Squibb, Daiichi Sankyo, Lilly, Novartis, MacroGenics, MSD, Pfizer, Puma Biotechnology, and Roche. Simona Borstnar has received honoraria for consulting or advisory roles from Amgen, AstraZeneca, Lilly, Novartis, Pfizer, and Roche; honoraria for participation in speakers’ bureaux from Amgen, Lilly, Krka, Novartis, Pfizer, and Roche. Mario Campone has received honoraria for consulting or advisory roles from AstraZeneca, Novartis, AbbVie, Sanofi, Lilly, Pfizer, Sandoz, Servier, Accord, and Pierre Fabre; honoraria from Novartis and Lilly; honoraria for participation in speakers’ bureaux from Novartis and Lilly; reimbursement for travel, accommodations, and expenses from Pfizer, Roche, and AstraZeneca. Ellen Warner has received reimbursement for travel, accommodations, or expenses from Novartis. Javier Salvador Bofill has received honoraria for consulting or advisory roles from Roche, Novartis, Pfizer, and MSD; honoraria for participation in speakers’ bureaux from Novartis and Roche; honoraria from Novartis, Pfizer, and Roche; reimbursement for travel, accommodations, or expenses from Roche, Novartis, and Pfizer. William Jacot has received honoraria for consulting or advisory roles from AstraZeneca, Eisai, Lilly France, MSD, Novartis, Pfizer, and Roche; research funding from AstraZeneca; reimbursement for travel, accommodations, or expenses from AstraZeneca, Chugai Pharma, Eisai, GlaxoSmithKline, and Lilly France. Susan Dent has received honoraria for consulting or advisory roles from Novartis, Pfizer, and Roche; honoraria from Eli Lilly, Novartis, and Roche; research funding from Hoffman La-Roche, Novartis, and Pfizer; reimbursement for travel, accommodations, or expenses from AstraZeneca. Miguel Martin has received honoraria for consulting or advisory roles from Amgen, AstraZeneca, Eli Lilly, Novartis, Pfizer, PharmaMar, Puma Biotechnology, Roche/Genentech, and Taiho Oncology; honoraria for participation in speakers’ bureaux from AstraZeneca, Amgen, Roche/Genentech, Novartis, and Pfizer; research funding from Roche and Novartis. Alistair Ring has received honoraria for consulting or advisory roles from Pfizer and Roche; honoraria from AstraZeneca, Lilly, Novartis, Pfizer, and Roche; research funding from AstraZeneca. Paul Cottu has received honoraria for consulting or advisory roles from Novartis and Pfizer; research funding from AstraZeneca, Genentech/Roche, Novartis, Pierre Fabre, and Pfizer; reimbursement for travel, accommodations, or expenses from AstraZeneca, NanoString Technologies, Novartis, Pfizer, and Roche. Janice Lu has received honoraria for consulting or advisory roles from Pfizer, Novartis, and Puma. Eva Ciruelos has received honoraria for consulting or advisory roles from Pfizer, Novartis, Lilly, and Roche; honoraria from Pfizer, Novartis, Lilly, and Roche; honoraria for participation in speakers’ bureaux from Pfizer, Novartis, Lilly, and Roche; reimbursement for travel, accommodations, or expenses from Pfizer and Roche. Hamdy A. Azim has received honoraria for consulting or advisory roles from ASZ, Bristol-Myers Squibb, Janssen, MSD, Novartis, Pfizer, Amgen, Lilly, and Roche; employment by Innate France (immediate family member); honoraria from Amgen, ASZ, Bristol-Myers Squibb, MSD, Novartis, Pfizer, and Roche; research funding from Pfizer. Sanjoy Chatterjee has received honoraria for consulting or advisory roles from Tata Medical Center. Katie Zhou is employed by Novartis. Jiwen Wu is employed by Novartis. Lakshmi Menon-Singh is employed by Novartis. Claudio Zamagni has received honoraria for consulting or advisory roles from AstraZeneca, Eisai, Novartis, Pfizer, PharmaMar, Pierre Fabre, and Roche; research funding from AbbVie, Array BioPharma, AstraZeneca, Celgene, Medivation, Morphotek, Novartis, Pfizer, Roche, and Roche/Genentech; reimbursement for travel, accommodations, or expenses from Celgene, Novartis, Pierre Fabre, and Roche.
Ethical approval
The study was designed, implemented, and reported in accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Harmonized Tripartite Guidelines for Good Clinical Practice, applicable local regulations, and the ethical principles laid down in the Declaration of Helsinki. The protocol and informed consent form were reviewed and approved by a properly constituted Institutional Review Board/Independent Ethics Committee/ Research Ethics Board before study commencement. A steering committee oversaw the trial conduct, as per the approved protocol. Representatives of the trial sponsor, Novartis Pharmaceuticals (East Hanover, NJ), collected and analyzed the data.
Consent to participate
Written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Laurentiis, M., Borstnar, S., Campone, M. et al. Full population results from the core phase of CompLEEment-1, a phase 3b study of ribociclib plus letrozole as first-line therapy for advanced breast cancer in an expanded population. Breast Cancer Res Treat 189, 689–699 (2021). https://doi.org/10.1007/s10549-021-06334-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-021-06334-0