
Abstract
Purpose
Gene expression (GE) profiling for breast cancer classification and prognostication has become increasingly used in clinical diagnostics. GE profiling requires a reasonable tumor cell percentage and high-quality RNA. As a consequence, a certain amount of samples drop out. If tumor characteristics are different between samples included and excluded from GE profiling, this can lead to bias. Therefore, we assessed whether patient and tumor characteristics differ between tumors suitable or unsuitable for generating GE profiles in breast cancer.
Methods
In a consecutive cohort of 738 breast cancer patients who received neoadjuvant chemotherapy at the Netherlands Cancer Institute, GE profiling was performed. We compared tumor characteristics and treatment outcome between patients included and excluded from GE profiling. Results were validated in an independent cohort of 812 patients treated with primary surgery.
Results
GE analysis could be performed in 53% of the samples. Patients with tumor GE profiles more often had high-grade tumors [odds ratio 2.57 (95%CI 1.77–3.72), p < 0.001] and were more often lymph node positive [odds ratio 1.50 (95%CI 1.03–2.19), p = 0.035] compared to the group for which GE profiling was not possible. In the validation cohort, tumors suitable for gene expression analysis were more often high grade.
Conclusions
In our gene expression studies, tumors suitable for GE profiling had more often an unfavorable prognostic profile. Due to selection of samples with a high tumor percentage, we automatically select for tumors with specific features, i.e., tumors with a higher grade and lymph node involvement. It is important to be aware of this phenomenon when performing gene expression analysis in a research or clinical context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DNA microarray analyses (e.g., tiling arrays, mRNA arrays, and direct sequencing of complementary DNA) have significantly advanced our understanding of breast cancer. They showed that breast cancer is not a single disease with variable morphologic features, but a group of molecular distinct neoplasms [1]. Furthermore, in certain clinical settings it can help to determine whether or not adjuvant chemotherapy is justified [2].
Several assays, resulting in risk scores, have been developed and are partially commercially and partially clinically available. The 21-gene Recurrence Score (OncotypeDx assay, Genome Health inc, Redwood city, CA) [3], the Amsterdam 70-gene profile, commercially known as the Mammaprint (Agendia, Huntington Beach, CA) assay [4] and the Risk of Recurrence (ROR) score, derived from Predictor Analysis of Microarray 50 (PAM50) [5], are mostly used. Reliable results of the assays require a good quality tumor sample with high cellularity. To illustrate this, Elloumi et al. [6] revealed a systematic bias when too much normal tissue was present in a tumor sample. However, tumor percentage is also dependent on tumor morphology. For example, a tumor with solid growth more easily reaches a high tumor percentage than a tumor with glandular or lobular growth (a feature important for grade). Furthermore, the presence of sclerosis as well as stromal and inflammatory cells can reduce the tumor cell percentage substantially. Another feature that influences the ability to perform gene expression analysis is the RNA quality. Pre-analytical factors such as time to fixation, fixation duration, and storage temperature have an impact on the RNA quality [7].
Summarizing the above, a high tumor percentage and good quality RNA are prerequisites for successful gene expression analysis. These requirements lead to the dropout of samples not fulfilling these criteria. As a higher histological grade is associated with a higher tumor cellularity, gene expression analysis might be more successful in high-grade tumors. To assess if indeed clinico-pathological variables are associated with successful gene expression analysis, we compared clinical and tumor characteristics of tumors suitable and unsuitable for gene expression analysis in two large (neo) adjuvant-treated patient cohorts.
Materials and methods
Patient selection
Tissue samples of patients treated in the neoadjuvant setting for breast cancer were collected at the Netherlands Cancer Institute (NKI) between 2004 and 2012. For participation in the neoadjuvant program, the tumor diameter should exceed 3 cm or axillary lymph node metastasis should be proven by fine-needle aspiration. Part of these patients participated in two ongoing clinical trials of which details have been described previously [8]. Both studies were approved by the ethical committee and informed consent for gene expression analysis was obtained for all included patients. At least two tumor biopsies were taken under ultrasound guidance, using a 14G core needle to assure sufficient tissue for both adequate diagnostics as well as for research purposes. To facilitate such analyses, at least one biopsy was snap frozen in liquid nitrogen and stored at − 80 degrees.
Pathology
Paraffin-embedded sections were all stained by a hematoxylin and eosin (H&E) stain and reviewed by a consultant breast pathologist (JW) for immunohistochemically assessment and histological classification (including subtype and grade) on biopsy material (of which the details are described previously) [9]. In brief, samples were scored as positive for oestrogen receptor (ER) and progesterone receptor (PR) if at least 10% of the tumor cells showed nuclear staining. HER2 (or ERBB2) was scored as positive when there was strong membranous staining in more than 30% of the tumor cells (3+) by immunohistochemistry or if chromosome in situ hybridization (CISH) revealed amplification. Percentage nuclear staining of tumor cells in Ki67 (MIB1) was scored as a marker for proliferation. Chemotherapy response was determined by pathological examination of resection specimens. Pathological complete remission (pCR) was defined as the absence of invasive tumor in both the breast and axillary lymph nodes after neoadjuvant chemotherapy.
Imaging data
For a subset of the patients, detailed imaging data were available. A dedicated breast radiologist (CL) assessed according to BIRADS lexicon [10] whether pre-treatment MRIs showed the tumor to be either mass-like, or non-mass like. For analysis purposes, these two categories were used. Metabolic activity was assessed using baseline 18F-fluorodeoxyglucose (FDG) positron emission tomography combined with computed tomography (PET/CT) scans. FDG uptake was quantified using maximum standardized uptake values (SUVmax) measured with Osirix DICOM viewer (Pixmeo SARL, Geneva, Swiss).
Gene expression assay and tumor percentage
mRNA was isolated from the frozen material as described previously [9]. Briefly, a 5-micrometer section of the biopsy was H&E stained. A pathologist evaluated if the overall tissue quality of the frozen biopsy was sufficient for further analysis (i.e., samples dropped out if the biopsy was too small, too fatty or in the absence of invasive tumor). The pathologist also estimated the tumor percentage and only the samples with a tumor percentage ≥ 50% were selected for microarray analysis. Gene expression analysis was only performed if the RNA integrity number ≥ 6.5 and the quantity ≥ 3 µg. Samples obtained between 2004 and 2010 were analyzed using Illumina microarray analysis (WG6 v3 microarray chips); RNAseq was performed on the samples from 2011 to 2012.
Validation cohort
As a validation, an independent cohort obtained from the microarRAy PrognoSTics in Breast cancER (RASTER) study was used. Study design is described before [11]. In short, 812 women were enrolled in 16 hospitals in The Netherlands after having given informed consent. Patients received surgery (mastectomy or breast conserving surgery) as primary treatment. Within 1 h after surgery, a tumor sample was procured at the pathology department of the participating hospitals and sent to Netherlands Cancer institute by mail. After samples were received at the Netherlands Cancer Institute they were snap frozen at − 70 degrees. Pathologists analyzed paraffin-embedded tumor samples of the validation dataset at the pathology department of the participating hospitals. Histological tumor grade according Elston and Ellis, ER status, PR status, and ERBB2 status were established by each hospital according to locally used methods [11]. Frozen sections of the tumor samples of the validation set were obtained and stained with H&E stain, and subsequently analyzed by an experienced breast pathologist. Eligible samples had to contain ≥ 50% tumor cells. Agendia Laboratories performed the microarray analysis using the Mammaprint (Agilent microarray, Santa Clara USA) [11].
Data analyses and statistics
The variables age, histologic subtype of tumor, grade, T-stage, N-stage, and response (pathological complete remission (pCR) of breast and axilla) were compared between samples suitable or unsuitable for gene expression analysis. The χ 2 (Spearman) was used to compare dichotomized variables. We also assessed differences in clinical characteristics for each exclusion criteria as described above, i.e., tissue quality of the frozen biopsy, tumor cell percentage, and RNA quantity as well as quality. Multivariate logistic regression analysis was performed to assess the independent association of various clinical variables with the ability to perform GE analysis. Recurrence-free survival was assessed with Kaplan–Meier plots and the log rank test. A cox proportional hazard model was built to assess if the ability to perform GE analysis was independently associated with recurrence-free survival. The SPSS Package 23.0 was used for statistical analyses and p values (two-sided) < 0.05 were considered statistically significant. This study was designed according to the Reporting recommendations for tumor MARKER prognostic studies (REMARK) guidelines [12].
For the validation dataset, the variables age, histologic type of tumor, subtype, histological grade, and T-stage were compared between tumor samples suitable or ineligible for gene expression analysis. Statistical analysis was performed as described above.
Results
Patient selection in cohort
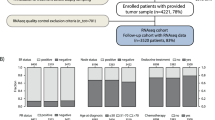
A total of 738 breast cancer patients were treated in the NKI with neoadjuvant chemotherapy between 2004 and 2012. From 665 patients, a frozen biopsy was available. Seventy-seven tissue samples were not processed because the biopsies were too small or too fatty, or did not contain invasive tumor. Of the remaining 587 tissue specimens, 461 had a tumor percentage of more than 50%; 391 of these samples met the criteria for sufficient RNA quality and quantity, allowing gene expression analysis (53% of the total cohort). Figure 1 shows the sample selection flow diagram.

Flow diagram of included patients. Flow diagram of included patients and in- and exclusion steps of the fresh frozen biopsies for gene expression studies. TP denotes tumor percentage, DCIS ductal carcinoma in situ, RIN RNA integrity number
Association with clinical characteristics
Comparisons of baseline characteristics between the tumors for which a gene expression (GE) profile could be generated (GE+ ; n = 391) and the tumors for which this was not possible (GE− ; n = 347) are shown in Table 1. GE + tumors were more often high grade and had a higher SUV max value than GE− tumors. When we stratify for subtype, the effect of grade is still significant in the ER+HER2− and in the triple-negative subgroup (see Supplementary, Table 1). In the ER+HER2− subgroup, these samples also have a higher SUV max value. Multivariate analysis shows that a high tumor grade and positive lymph node status are independently associated with GE + tumors (Table 2).
To investigate the influence of the various steps of sample selection for gene expression analysis in more detail, we compared the clinical variables between in- and excluded samples after each selection step (Table 3). Interestingly, in every selection step, we enrich for high-grade tumors: a high tumor grade is associated with larger biopsies, higher tumor percentage and high quality and quantity RNA. In addition, high quality and sufficient quantity of RNA is more often found in HER2+ tumors and node-positive tumors.
Association with treatment response and survival
Chemotherapy response was not significantly different between GE + and GE − tumors. However, we observed that tumors with high tissue quality of frozen biopsies more often achieved a pathological compete response (pCR) after treatment than samples with poor quality biopsies (p = 0.02; Table 3). We did not observe a significant difference in recurrence-free survival between samples included and excluded in gene expression analysis, although a trend was visible in triple-negative patients (Fig. 2). A cox proportional hazard model did not indicate GE+ as a variable associated with survival (Supplementary, Table 2).

Kaplan–Meier curves of recurrence-free survival. Kaplan–Meier curves of recurrence-free survival to compare patients with and without gene expression profiles, stratified by subtype. GE denoted gene expression
Results in the validation dataset
The RASTER data were used to validate our observations. This set includes 812 breast cancer patients enrolled between 2004 and 2006 (see Methods). Because node positive patients were excluded in this study for clinical reasons and therefore a gene expression profile was not performed, we analyzed the samples of node-negative patients (n = 585, see Supplementary Fig. 1, for a flowchart). Therefore, we could not validate our association with nodal status in this set; however, most other clinical variables were available. Of these samples, 27% dropped out because of incorrect procedure or sample failure. Similar to the observations in the neoadjuvant cohort, gene expression profiling was more often possible in high-grade tumors (borderline significant; p = 0.05, Table 4).
Discussion
In this study, we showed that patients of whom gene expression data were obtained had more often high-grade tumors and lymph node metastasis, features associated with a worse prognosis. It is important to acknowledge that, due to the selection of tumors with good quality samples and a high cellularity for gene expression studies, we select for a certain subgroup of tumors. Most likely, this selection bias is present in the majority of published gene expression studies for breast cancer.
Our findings are indeed in line with published literature. Cremoux et al. [13] studied (pre-) analytical steps in tissue handling by comparing different institutes. As in our study, they found that tumors suitable for gene expression profiling were more often high grade and of ductal subtype. Mook et al. observed a 17% dropout and concluded that the rejected samples were obtained from slightly smaller tumors [14]. Also Goetz et al. observed that women without expression data had more often a small tumor [15]. Together with the results of our validation dataset, there is substantial evidence towards the selection of larger and more aggressive tumors for gene expression studies.
In a systematic literature search that we performed prior to this study, finally resulting in 110 articles, 39% of the studies were indistinct about exclusion criteria and associated dropout rates, indicating unawareness. The remaining 61% did mention about exclusion criteria and numbers. These studies show a variety of dropout rates (1–83%, average 21%). Most gene expression profiles had been developed on frozen sample collections available at the biobank of the respective institute. These samples are a selection of relatively large and easily accessible tumors. Also, only samples that met the strict criteria of tumor RNA quality and quantity were used. Nowadays FFPE material from all laboratories (both biopsies and resection material), with very different protocols, can be measured by commercial available platforms, such as Mammaprint and OncotypeDx, and tumor percentage can be as low as 30% [16]. This is possible due to advances in the technique. However, we should be aware that such assays were originally not developed on samples with comparable characteristics, and validation on small samples with lower tumor cell percentage is warranted. In addition, also for research purposes, it is important to acknowledge that due to analytical requirements high-grade tumors might be overrepresented in GE datasets.
This study has some limitations. First, fresh frozen tumor samples were used. In general, it is more difficult to obtain fresh frozen material than FFPE tumor material, resulting in a higher dropout rate. Second, this study was done on pre-treatment biopsies, which yield smaller quantities of tumor material than resection specimens. Third, this study was performed in the neoadjuvant setting, which results in the selection of locally advanced tumors. Consequently, we could not look at stage I or stage IV tumors. Strong points of our study are that samples were obtained from a consecutive cohort of neoadjuvant treated patients, and not on a highly selected clinical trial population. Furthermore, we had an independent cohort for validation purposes that corroborated our findings. Although there were some differences in the way the samples were collected (resections versus biopsies, mailed transport versus snap frozen), the validation cohort consisted of early stage breast cancer samples and had information on grade, enabling us to validate our main finding in an independent cohort. Finally, all samples were from one institute and handled by one dedicated technician to preclude variability in centre or in lab handling.
In conclusion, we showed that breast cancers for which gene expression data were successfully obtained were associated with a higher grade and with lymph node metastasis, due to the selection of samples with a high tumor percentage and good quality RNA. These tumors have, on average, a more aggressive phenotype and a relatively poor prognosis. In general, when interpreting test results, it is important to realize that patient populations for which GE profiles are used, often differ substantially from the ones in which they were originally developed, particularly when using a development cohort consisting of frozen tumor tissue and a test cohort consisting of FFPE samples. At this point, it is uncertain what the impact might be on treatment decisions in the clinic.
References
Sotiriou C, Pusztai L (2009) Gene-expression signatures in breast cancer. N Engl J Med 360(8):790–800. https://doi.org/10.1056/NEJMra0801289
Ward S, Scope A, Rafia R, Pandor A, Harnan S, Evans P, Wyld L (2013) Gene expression profiling and expanded immunohistochemistry tests to guide the use of adjuvant chemotherapy in breast cancer management: a systematic review and cost-effectiveness analysis. Health Technol Assess 17(44):1–302. https://doi.org/10.3310/hta17440
Mamounas EP, Tang G, Fisher B, Paik S, Shak S, Costantino JP, Watson D, Geyer CE Jr, Wickerham DL, Wolmark N (2010) Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor-positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol 28(10):1677–1683. https://doi.org/10.1200/JCO.2009.23.7610
van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871):530–536. https://doi.org/10.1038/415530a
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron J, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167. https://doi.org/10.1200/JCO.2008.18.1370
Elloumi F, Hu Z, Li Y, Parker JS, Gulley ML, Amos KD, Troester MA (2011) Systematic bias in genomic classification due to contaminating non-neoplastic tissue in breast tumor samples. BMC Med Genom 4:54. https://doi.org/10.1186/1755-8794-4-54
Bass BP, Engel KB, Greytak SR, Moore HM (2014) A review of preanalytical factors affecting molecular, protein, and morphological analysis of formalin-fixed, paraffin-embedded (FFPE) tissue: how well do you know your FFPE specimen? Arch Pathol Lab Med 138(11):1520–1530. https://doi.org/10.5858/arpa.2013-0691-RA
Lips EH, Mulder L, de Ronde JJ, Mandjes IA, Koolen BB, Wessels LF, Rodenhuis S, Wesseling J (2013) Breast cancer subtyping by immunohistochemistry and histological grade outperforms breast cancer intrinsic subtypes in predicting neoadjuvant chemotherapy response. Breast Cancer Res Treat 140(1):63–71. https://doi.org/10.1007/s10549-013-2620-0
Lips EH, Mulder L, de Ronde JJ, Mandjes IAM, Vincent A, Vrancken Peeters MTFD, Nederlof PM, Wesseling J, Rodenhuis S (2012) Neoadjuvant chemotherapy in ER + HER2 − breast cancer: response prediction based on immunohistochemical and molecular characteristics. Breast Cancer Res Treat 131(3):827–836. https://doi.org/10.1007/s10549-011-1488-0
Morris EA, Comstock CE, Lee CH (2013) ACR BI-RADS® Magnetic resonance imaging. ACR BI-RADS® Atlas, Breast imaging reporting and data system. American College of Radiology, Reston, VA
Bueno-de-Mesquita JM, van Harten WH, Retel VP, van’t Veer LJ, van Dam FS, Karsenberg K, Douma KF, van Tinteren H, Peterse JL, Wesseling J, Wu TS (2007) Use of 70-gene signature to predict prognosis of patients with node-negative breast cancer: a prospective community-based feasibility study (RASTER). Lancet oncol. 8(12):1079–1087
Altman DG, McShane LM, Sauerbrei W, Taube SE (2012) Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. PLoS medicine 9(5):e1001216. https://doi.org/10.1371/journal.pmed.1001216
de Cremoux P, Valet F, Gentien D, Lehmann-Che J, Scott V, Tran-Perennou C, Barbaroux C, Servant N, Vacher S, Sigal-Zafrani B, Mathieu MC, Bertheau P, Guinebretiere JM, Asselain B, Marty M, Spyratos F (2011) Importance of pre-analytical steps for transcriptome and RT-qPCR analyses in the context of the phase II randomised multicentre trial REMAGUS02 of neoadjuvant chemotherapy in breast cancer patients. BMC Cancer 11:215. https://doi.org/10.1186/1471-2407-11-215
Mook S, Schmidt MK, Viale G, Pruneri G, Eekhout I, Floore A, Glas AM, Bogaerts J, Cardoso F, Piccart-Gebhart MJ, Rutgers ET, Van’t Veer LJ (2009) The 70-gene prognosis-signature predicts disease outcome in breast cancer patients with 1–3 positive lymph nodes in an independent validation study. Breast Cancer Res Treat 116(2):295–302. https://doi.org/10.1007/s10549-008-0130-2
Goetz MP, Suman VJ, Ingle JN, Nibbe AM, Visscher DW, Reynolds CA, Lingle WL, Erlander M, Ma XJ, Sgroi DC, Perez EA, Couch FJ (2006) A two-gene expression ratio of homeobox 13 and interleukin-17B receptor for prediction of recurrence and survival in women receiving adjuvant tamoxifen. Clin Cancer Res 12(7 Pt 1):2080–2087. https://doi.org/10.1158/1078-0432.ccr-05-1263
Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, Cronin M, Baehner FL, Watson D, Bryant J, Costantino JP, Geyer CE Jr, Wickerham DL, Wolmark N (2006) Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 24(23):3726–3734. https://doi.org/10.1200/JCO.2005.04.7985
Acknowledgements
We would like to acknowledge Kenneth Gilhuijs and Kenneth Pengel for providing imaging data of the tumors. We would like to acknowledge the NKI-AVL Core facility Molecular pathology & biobanking (CFMPB) for supplying NKI-AVL biobank material and laboratory support.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
EL, JW designed the study. MS, AM did the literature search. LW, SR, GS, CD provided clinical data. CL and BP provided imaging data. EL, LM prepared the samples. EL, JW, MS, AM interpreted the data and wrote the manuscript. AM, MS analyzed the data. MS, AM, LM, MH, SR, GS, JW, EL discussed the data. All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and informed consent
Specific approval and informed consent for the current study was not required, because the current study comprises a retrospective analysis of residual tissue samples. According to the policy of The Netherlands Cancer Institute, residual tissue may be used for scientific purposes unless a patient explicitly objects.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Seijen, M., Mooyaart, A.L., Mulder, L. et al. Enrichment of high-grade tumors in breast cancer gene expression studies. Breast Cancer Res Treat 168, 327–335 (2018). https://doi.org/10.1007/s10549-017-4622-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4622-9