
Abstract
Mitochondrial diseases are a group of genetic disorders leading to the dysfunction of mitochondrial energy metabolism pathways. We aimed to assess the clinical phenotype and the biochemical cerebrospinal fluid (CSF) biogenic amine profiles of patients with different diagnoses of genetic mitochondrial diseases. We recruited 29 patients with genetically confirmed mitochondrial diseases harboring mutations in either nuclear or mitochondrial DNA (mtDNA) genes. Signs and symptoms of impaired neurotransmission and neuroradiological data were recorded. CSF monoamines, pterins, and 5-methyltetrahydrofolate (5MTHF) concentrations were analyzed using high-performance liquid chromatography with electrochemical and fluorescence detection procedures. The mtDNA mutations were studied by Sanger sequencing, Southern blot, and real-time PCR, and nuclear DNA was assessed either by Sanger or next-generation sequencing. Five out of 29 cases showed predominant dopaminergic signs not attributable to basal ganglia involvement, harboring mutations in different nuclear genes. A chi-square test showed a statistically significant association between high homovanillic acid (HVA) values and low CSF 5-MTHF values (chi-square = 10.916; p = 0.001). Seven out of the eight patients with high CSF HVA values showed cerebral folate deficiency. Five of them harbored mtDNA deletions associated with Kearns-Sayre syndrome (KSS), one had a mitochondrial point mutation at the mtDNA ATPase6 gene, and one had a POLG mutation. In conclusion, dopamine deficiency clinical signs were present in some patients with mitochondrial diseases with different genetic backgrounds. High CSF HVA values, together with a severe cerebral folate deficiency, were observed in KSS patients and in other mtDNA mutation syndromes.



Similar content being viewed by others
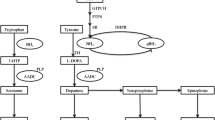

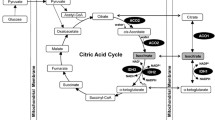
References
Alebouyeh M, Takeda M, Onozato ML et al (2003) Expression of human organic anion transporters in the choroid plexus and their interactions with neurotransmitter metabolites. J Pharmacol Sci 93:430–436
Allen RJ, DiMauro S, Coulter DL, Papadimitriou A, Rothenberg SP (1983) Kearns-Sayre syndrome with reduced plasma and cerebrospinal fluid folate. Ann Neurol 13:679–682
Asencio C, Rodríguez-Hernandez MA et al (2016) Severe encephalopathy associated to pyruvate dehydrogenase mutations and unbalanced coenzyme Q10 content. Eur J Hum Genet 24:367–372
Aylett SB, Neergheen V, Hargreaves IP et al (2013) Levels of 5-methyltetrahydrofolate and ascorbic acid in cerebrospinal fluid are correlated: implications for the accelerated degradation of folate by reactive oxygen species. Neurochem Int 63:750–755
Brito S, Thompson K, Campistol J et al (2015) Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front Genet 6:102. https://doi.org/10.3389/fgene.2015.00102.
Carlsson A (2001) A paradigm shift in brain research. Science 294:1021–1024
De Grandis E, Serrano M, Pérez-Dueñas B et al (2010) Cerebrospinal fluid alterations of the serotonin product, 5- hydroxyindolacetic acid, in neurological disorders. J Inherit Metab Dis 33:803–809
Dougados M, Zittoun J, Laplane D, Castaigne P (1983) Folate metabolism disorder in Kearns-Sayre syndrome. Ann Neurol 13:687
García-Cazorla A, Serrano M, Pérez-Dueñas B et al (2007) Secondary abnormalities of neurotransmitters in infants with neurological disorders. Dev Med Child Neurol 49:740–744
Garcia-Cazorla A, Duarte S, Serrano M et al (2008a) Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion 8:273–278
Garcia-Cazorla A, Quadros EV, Nascimento A et al (2008b) Mitochondrial diseases associated with cerebral folate deficiency. Neurology 70:1360–1362
Ghaoui R, Sue CM (2018) Movement disorders in mitochondrial disease. J Neurol 265:1230–1240
Grapp M, Wrede A, Schweizer M et al (2013) Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat Commun 4:2123
Haddad D, Nakamura K (2015) Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett 589:3702–3713
Hasselmann O, Blau N, Ramaekers VT, Quadros EV, Sequeira JM, Weissert M (2010) Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet Metab 99:58–61
Horvath GA, Demos M, Shyr C et al (2016) Secondary neurotransmitter deficiencies in epilepsy caused by voltage-gated sodium channelopathies: a potential treatment target? Mol Genet Metab 117:42–48
Hyland K, Surtees RA, Heales SJ, Bowron A, Howells DW, Smith I (1993) Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr Res 34:10–14
Invernizzi F, Varanese S, Thomas A, Carrara F, Onofrj M, Zeviani M (2008) Two novel POLG1 mutations in a patient with progressive external ophthalmoplegia, levodopa-responsive pseudo-orthostatic tremor and parkinsonism. Neuromuscul Disord 18:460–464
Kurian MA, Gissen P, Smith M, Heales S Jr, Clayton PT (2011) The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 10:721–733
Kuster A, Arnoux JB, Barth M et al (2018) Diagnostic approach to neurotransmitter monoamine disorders: experience from clinical, biochemical, and genetic profiles. J Inherit Metab Dis 41:129–139
Ly CV, Verstreken P (2006) Mitochondria at the synapse. Neuroscientist 12:291–299
Marecos C, Ng J, Kurian MA (2014) What is new for monoamine neurotransmitter disorders? J Inherit Metab Dis 37:619–626
Miguel R, Gago MF, Martins J (2014) POLG1-related levodopa-responsive parkinsonism. Clin Neurol Neurosurg 126:47–54
Molero-Luis M, Fernández-Ureña S, Jordán I et al (2013a) Cerebrospinal fluid neopterin analysis in neuropediatric patients: establishment of a new cut off-value for the identification of inflammatory-immune mediated processes. PLoS One 8:e83237
Molero-Luis M, Serrano M, Ormazábal A et al (2013b) Homovanillic acid in cerebrospinal fluid of 1388 children with neurological disorders. Dev Med Child Neurol 55:559–566
Montiel-Sosa JF, Herrero MD, Munoz M de L et al (2013) Phylogenetic analysis of mitochondrial DNA in a patient with Kearns-Sayre syndrome containing a novel 7629-bp deletion. Mitochondrial DNA 24:420–431
Moran MM, Allen NM, Treacy EP, King MD (2011) “Stiff neonate” with mitochondrial DNA depletion and secondary neurotransmitter defects. Pediatr Neurol 45:403–405
Mori S, Takanaga H, Ohtsuki S et al (2003) Rat organic anion transporter 3 (rOAT3) is responsible for brain-to-blood efflux of homovanillic acid at the abluminal membrane of brain capillary endothelial cells. J Cereb Blood Flow Metab 23:432–440
Moy LY, Wang SP, Sonsalla PK (2007) Mitochondrial stress-induced dopamine efflux and neuronal damage by malonate involves the dopamine transporter. J Pharmacol Exp Ther 320:747–756
Murthy VN, De Camilli P (2003) Cell biology of the presynaptic terminal. Annu Rev Neurosci 26:701–728
Ng J, Papandreou A, Heales SJ, Kurian MA (2015) Monoamine neurotransmitter disorders–clinical advances and future perspectives. Nat Rev Neurol 11:567–584
O'Callaghan MM, Emperador S, Pineda M et al (2015) Mutation loads in different tissues from six pathogenic mtDNA point mutations. Mitochondrion 22:17–22
Orešković D, Radoš M, Klarica M (2017) Role of choroid plexus in cerebrospinal fluid hydrodynamics. Neuroscience 354:69–87
Ormazabal A, García-Cazorla A, Fernández Y, Fernández-Alvarez E, Campistol J, Artuch R (2005) HPLC with electrochemical and fluorescence detection procedures for the diagnosis of inborn errors of biogenic amines and pterins. J Neurosci Methods 142:153–158
Ormazabal A, García-Cazorla A, Pérez-Dueñas B et al (2006) Determination of 5-methyltetrahydrofolate in cerebrospinal fluid of paediatric patients: reference values for a paediatric population. Clin Chim Acta 371:159–162
Ortigoza-Escobar JD, Molero-Luis M, Arias A et al (2016) Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: a treatable cause of Leigh syndrome. Brain 139:31–38
Pearl PL, Capp PK, Novotny EJ, Gibson KM (2005) Inherited disorders of neurotransmitters in children and adults. Clin Biochem 38:1051–1058
Pérez-Dueñas B, Ormazábal A, Toma C et al (2011) Cerebral folate deficiency syndromes in childhood: clinical, analytical, and etiologic aspects. Arch Neurol 68:615–621
Pineda M, Ormazabal A, Lopez-Gallardo E et al (2006) Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann Neurol 59:394–398
Ramaekers VT, Blau N (2004) Cerebral folate deficiency. Dev Med Child Neurol 46:843–851
Ramaekers VT, Weis J, Sequeira JM, Quadros EV, Blau N (2007) Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency. Neuropediatrics 38:184–187
Rodan LH, Gibson KM, Pearl PL (2015) Clinical use of CSF neurotransmitters. Pediatr Neurol 53:277–286
Serrano M, García-Silva MT, Martin-Hernandez E et al (2010) Kearns-Sayre syndrome: cerebral folate deficiency, MRI findings and new cerebrospinal fluid biochemical features. Mitochondrion 10:429–432
Spector R (2010) Nature and consequences of mammalian brain and CSF efflux transporters: four decades of progress. J Neurochem 112:13–23
Spector R, Johanson CE (2010) Choroid plexus failure in the Kearns-Sayre syndrome. Cerebrospinal Fluid Res 7:14
Tanji K, Schon EA, DiMauro S, Bonilla E (2000) Kearns-Sayre syndrome: oncocytic transformation of choroid plexus epithelium. J Neurol Sci 178:29–36
Tondo M, Málaga I, O'Callaghan M et al (2011) Biochemical parameters to assess choroid plexus dysfunction in Kearns-Sayre syndrome patients. Mitochondrion 11:867–870
Tzoulis C, Tran GT, Schwarzlmüller T et al (2013) Severe nigrostriatal degeneration without clinical parkinsonism in patients with polymerase gamma mutations. Brain 136:2393–2404
Tzoulis C, Schwarzlmüller T, Biermann M, Haugarvoll K, Bindoff LA (2016) Mitochondrial DNA homeostasis is essential for nigrostriatal integrity. Mitochondrion 28:33–37
Van Der Heyden JC, Rotteveel JJ, Wevers RA et al (2003) Decreased homovanillic acid concentrations in cerebrospinal fluid in children without a known defect in dopamine metabolism. Eur J Paediatr Neurol 7:31–37
Yubero D, Brandi N, Ormazabal A et al (2016) Targeted next generation sequencing in patients with inborn errors of metabolism. PLoS One 11:e0156359
Acknowledgements
The Departments of Clinical Biochemistry and Genetics are part of the “Centre Daniel Bravo de Diagnòstic i Recerca en Malalties Minoritàries.” We are indebted to the Spanish Association of Mitochondrial Patients (AEPMI) and the Fundación Carolina Diaz-Mahou.
Funding
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII-FIS PI17/00109, PI17/00021 and PI15/01082), the FEDER Funding Program from the European Union, and CIBERER-ISCIII.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M. Batllori, M. Molero-Luis, A. Ormazabal, R. Montero, C. Sierra, A. Ribes, J. Montoya, E. Ruiz-Pesini, M. O’Callaghan, L. Pias, A. Nascimento, F. Palau, J. Armstrong, D. Yubero, J. D. Ortigoza-Escobar, A. García-Cazorla, and R. Artuch declare that they have no conflict of interest.
Additional information
Communicating Editor: Nenad Blau
Rights and permissions
About this article

Cite this article
Batllori, M., Molero-Luis, M., Ormazabal, A. et al. Cerebrospinal fluid monoamines, pterins, and folate in patients with mitochondrial diseases: systematic review and hospital experience. J Inherit Metab Dis 41, 1147–1158 (2018). https://doi.org/10.1007/s10545-018-0224-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-018-0224-x