
Abstract
Introduction
Currently, no therapies are available for Zellweger spectrum disorders (ZSDs), a group of genetic metabolic disorders characterised by a deficiency of functional peroxisomes. In a previous study, we showed that oral cholic acid (CA) treatment can suppress bile acid synthesis in ZSD patients and, thereby, decrease plasma levels of toxic C27-bile acid intermediates, one of the biochemical abnormalities in these patients. However, no effect on clinically relevant outcome measures could be observed after 9 months of CA treatment. It was noted that, in patients with advanced liver disease, caution is needed because of possible hepatotoxicity.
Methods
An extension study of the previously conducted pretest–posttest design study was conducted including 17 patients with a ZSD. All patients received oral CA for an additional period of 12 months, encompassing a total of 21 months of treatment. Multiple clinically relevant parameters and markers for bile acid synthesis were assessed after 15 and 21 months of treatment.
Results
Bile acid synthesis was still suppressed after 21 months of CA treatment, accompanied with reduced levels of C27-bile acid intermediates in plasma. These levels significantly increased again after discontinuation of CA. No significant changes were found in liver tests, liver elasticity, coagulation parameters, fat-soluble vitamin levels or body weight.
Conclusions
Although CA treatment did lead to reduced levels of toxic C27-bile acid intermediates in ZSD patients without severe liver fibrosis or cirrhosis, no improvement of clinically relevant parameters was observed after 21 months of treatment. We discuss the implications for CA therapy in ZSD based on these results.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
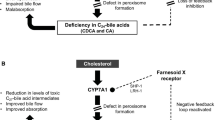
Zellweger spectrum disorders (ZSDs) are a subgroup of the peroxisome biogenesis disorders and are caused by biallelic mutations in one of the PEX genes, leading to a deficiency of functional peroxisomes. Since peroxisomes are responsible for a variety of metabolic functions, ZSDs are characterised by multiple biochemical abnormalities, including the accumulation of C27-bile acid intermediates [3α,7α-dihydroxycholestanoic acid (DHCA) and 3α,7α,12α-trihydroxycholestanoic acid (THCA)], very long-chain fatty acids (VLCFAs), branched-chain fatty acids like phytanic acid and pristanic acid, and plasmalogen deficiency (Wanders and Waterham 2006). Patients show a spectrum of clinical manifestations, including retinitis pigmentosa, sensorineural hearing loss, leukodystrophy and liver disease as key symptoms. Liver disease encompasses hepatomegaly with or without fibrosis or cirrhosis. Those with cirrhosis are at risk of complications, like portal hypertension, cholestasis, liver failure (Klouwer et al. 2015; Braverman et al. 2016) and hepatocellular carcinoma (unpublished observations). Signs of liver disease can be completely absent in ZSD patients with a relatively mild phenotype. It is still largely unknown which biochemical abnormalities, either alone or in combination, contribute to the individual clinical manifestations, although some correlations have been hypothesised (Braverman et al. 2013; Klouwer et al. 2015). The C27-bile acid intermediates are toxic and thought to contribute to the liver disease (Ferdinandusse et al. 2009; Wanders and Ferdinandusse 2012); however, a role for other biochemical abnormalities in this process cannot be ruled out.
Only symptomatic therapies are currently available for patients with ZSDs. Supplementation with the primary bile acid cholic acid (CA) was suggested to be a potential therapy for patients with a ZSD, since CA represses the first step of bile acid biosynthesis via the major pathway, thereby leading to reduced levels of toxic C27-bile acid intermediates (Setchell et al. 1992). Moreover, it was hypothesised that CA therapy, through expansion of the expected small bile acid pool in patients with ZSDs, would lead to improved solubilisation of dietary fats and fat-soluble vitamins, leading to a reduction of steatorrhoea, increased fat-soluble vitamin levels and improved growth.
Previously, we performed a systematic study of the effects of 9 months of oral CA treatment in 19 patients with a ZSD (Berendse et al. 2016b). Bile acid synthesis was, indeed, suppressed in most patients during treatment, and consequently led to decreased C27-bile acid intermediates in plasma and urine of those patients. However, despite the clear biochemical effects, no positive effects on clinically relevant parameters could be observed after 9 months of CA treatment. No changes were observed in fat-soluble vitamin levels, coagulation parameters or body weight. Plasma transaminases [alanine transaminase (ALT) and aspartate transaminase (AST)] and conjugated bilirubin levels did not improve, although it should be noted that most patients had normal levels at baseline. However, four patients with liver cirrhosis at baseline showed a profound increase in transaminases and bilirubin levels upon CA treatment, and discontinuation or dose reduction was necessary. It was, therefore, concluded that CA therapy can be potentially harmful for ZSD patients with advanced liver disease and that 9 months of treatment was most likely too short to conclude whether CA therapy may lead to a clinical benefit in ZSD patients with a relatively mild liver phenotype.
In this study, we investigated the effect of 21 months of CA treatment on biochemical parameters and clinically relevant outcome measures in 17 patients with a ZSD. In addition, three additional young patients were included in the initial 9 months treatment phase.
Methods
Study design
This study is an extension study of a previously conducted pretest–posttest design study in which 19 ZSD patients were treated with CA for 9 months after a run-in period of 2 years. The results of this study have been previously published (Berendse et al. 2016b). The initial treatment phase took place between April 2014 and January 2015 and was extended till January 2016 (trial registry: http://www.isrctn.com/ISRCTN96480891). The extension was approved by the Institutional Review Board of the Academic Medical Center (AMC) in Amsterdam. Individual written informed consent was obtained from the patients and/or the patient’s parents. Two additional study visits at 15 and 21 months after the start of CA treatment took place during the extended treatment phase. Patients were followed up in regular patient care after discontinuation of CA at the end of the study. Similar to the initial study, blood and urine for biochemical analyses were collected during both study visits and a physical examination was performed. Liver stiffness analyses were only performed at the end of the extended treatment phase.
The primary study objective was to assess whether the previously observed suppression of bile acid synthesis [as defined by the change in levels of the bile acid intermediate 7α-hydroxy-4-cholesten-3-one (C4) and the C27-bile acid intermediates DHCA and THCA in plasma, and the urinary occurrence of bile acid intermediates] was still present after 21 months of CA treatment. Fibroblast growth factor 19 (FGF19) is a bile acid-induced enterokine that negatively regulates bile acid synthesis by acting on the liver to downregulate cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme in de novo bile acid synthesis (Schaap et al. 2014), and was, therefore, measured to evaluate the effect of CA on bile acid metabolism.
The secondary study objectives were: (1) to determine whether suppression of bile acid synthesis during a period of 21 months leads to improved clinically relevant parameters (i.e. liver tests and protein synthesis, liver elasticity, fat-soluble vitamin levels and growth), (2) to monitor the possible side effects of CA treatment during a prolonged treatment phase (including: change in conjugated bilirubin and plasma transaminases ALT and AST) and (3) to determine whether patients who develop a worsening of liver tests upon CA can be identified before initiating CA treatment.
Patients
All ZSD patients who were included in the initial study and did not show a two-fold or more increase of conjugated bilirubin, ALT or AST levels upon CA treatment in the initial 9-month follow-up period were eligible for inclusion in the extension study. ZSD patients who were not included in the initial study or were born after April 2014 (i.e. the start of the 9 months treatment phase of the initial study), were included for 9 months of CA treatment when they met the previously described inclusion and exclusion criteria (Berendse et al. 2016b). The frequency and characteristics of the study visits for these patients were identical to those in the initial study. All patients attended the outpatient clinic of the AMC for study visits.
CA
The active pharmaceutical ingredient (API) of CA was provided by Asklepion Pharmaceuticals (since 2015: Retrophin, Inc., New York, NY, USA). Capsules were developed and manufactured by Tiofarma (Oud Beijerland, the Netherlands), according to the guidelines of Good Manufacturing Practice commissioned by the AMC pharmacy, resulting in two dosage forms; yellow 50-mg capsules and opaque 250-mg capsules. CA capsules of 50 and/or 250 mg were administered twice a day orally during or shortly before meals. In 17 patients, CA was administrated orally and in four out of 21 patients via a percutaneous endoscopic gastrostomy tube. When CA was administrated via a percutaneous endoscopic gastrostomy tube or when patients were unable to swallow capsules, the capsules were opened and their content mixed with water or food. The dosage of CA of an individual patient at the end of the initial study was maintained during the extension study. Patients who were not included in the initial study started with a total dosage of 15 mg/kg/day. The dosage was increased to 20 mg/kg/day in case C27-bile acid intermediates DHCA and/or THCA were still detectable in plasma and/or urine and when this patient experienced no side effects. The effect of the dose escalation was checked after 4 weeks. In case side effects occurred, particularly diarrhoea, vomiting or worsening of liver tests (defined as a two-fold or more increase in plasma transaminases or conjugated bilirubin from baseline), the dosage was reduced to 10 mg/kg/day. The CA treatment was discontinued in case the plasma transaminases and/or conjugated bilirubin levels did not decrease after dose reduction.
Biochemical analysis, liver stiffness measurements and physical examination
Plasma and urinary bile acids were measured as described (Bootsma et al. 1999). The lower detection limit of bile acid intermediates in this assay is 0.05 μmol/L. Urinary bile acids were measured qualitatively only, and comprised primary bile acids (conjugates), bile alcohols and C27-bile acid intermediates as described (Ferdinandusse and Houten 2006). Plasma FGF19 and FGF21 were determined using specific enzyme-linked immunosorbent assays (ELISAs; lower detection limit of 0.01 ng/mL for both assays) as described elsewhere (Schreuder et al. 2010; Koot et al. 2013). Plasma C4 was measured using liquid chromatography–mass spectrometry (LC-MS) after acetonitrile precipitation (Lenícek et al. 2016), with a lower detection limit of 1 ng/mL. Standard diagnostic assays were used to measure levels of conjugated bilirubin, plasma transaminases, fat-soluble vitamins, total cholesterol, albumin and coagulation factors [i.e. prothrombin time (PT), partial thromboplastin time (aPTT)].
Liver stiffness analyses were performed by a single trained observer using transient elastography (FibroScan®), according to the standard manufacturer instructions (Echosens, Paris, France). Severe liver fibrosis or cirrhosis was defined as a FibroScan® value ≥ 15.5 kPa (de Lédinghen et al. 2007).
All patients underwent a standard physical and neurological examination at each study visit. Weight was measured with a calibrated balance, and age- and gender-specific standard deviation (SD) scores were calculated using current Dutch reference values (Talma et al. 2010).
Statistical analysis
Statistical analyses were performed with GraphPad Prism software version 7.02. A Wilcoxon matched-pairs signed-rank sum test was used to evaluate effects (baseline vs. the follow-up time points) and a Mann–Whitney U-test was used to compare median levels at baseline between different subgroups of patients. A p-value of < 0.05 was considered statistically significant.
Results
The individual patient characteristics are presented in Table 1. The study numbers of the included patients correspond to the data from the initial paper (Berendse et al. 2016b). A total of 22 ZSD patients were included in the 9 months treatment phase (median age 9.5 years). Nineteen of these were previously reported (Berendse et al. 2016b). Seventeen completed the 21 months treatment phase (median age 15 years). Three patients were diagnosed with a ZSD after April 2014 and were only included for 9 months of CA treatment. Two patients were excluded during (patient #16 after 3 months) or after (patient #19 after 9 months) the initial 9 months treatment phase because of a persistent rise in levels of conjugated bilirubin and/or plasma transaminases. No patients were lost during follow-up and vitamins were supplemented at a fixed dose in all patients throughout the study. Six patients (#16–19, 21 and 22) in the 9 months treatment phase had severe liver fibrosis or cirrhosis at baseline. Two patients with severe liver fibrosis or cirrhosis completed 21 months of CA treatment.
Measurements of bile acid intermediates
The median plasma levels of the C27-bile acid intermediates DHCA and THCA significantly decreased after 1, 3, 9, 15 and 21 months of CA treatment compared to baseline (Fig. 1a, b). When excluding baseline data from the patients who did not complete the full duration of 21 months of treatment, the median levels after 15 and 21 months of treatment remained significantly lower compared to the median baseline levels (data not shown). No significant differences in the median plasma DHCA and THCA levels were observed at baseline and 2 years prior to treatment initiation (Fig. 1a, b). Six to 12 months after discontinuation of CA (i.e. end of the treatment phase), the median DHCA and THCA levels significantly increased and approached pre-treatment levels (Fig. 1c, d). Nine out of 21 patients had detectable C27-bile acid intermediates in urine at baseline (one missing value). For eight of these patients, follow-up urine analysis was available, revealing detectable C27-bile acid intermediates in two cases after 3 months of treatment. One of these patients was excluded afterwards (#16). The other patient (#19) had no detectable intermediates in urine after 9 months of treatment, but was also excluded from the study after that study visit due to signs of liver toxicity. The individual levels of the C27-bile acid intermediates in plasma and urine for each time point are presented in Supplementary Table 1.

a, b Tukey box plots showing the effect of oral cholic acid (CA) on plasma 3α,7α-dihydroxycholestanoic acid (DHCA) and 3α,7α,12α-trihydroxycholestanoic acid (THCA) after 1, 3, 9, 15 and 21 months of treatment. The control reference range for THCA is < 0.05–0.1 μmol/L and levels of DHCA are undetectable (< 0.05 μmol/L) in control individuals. c, d Tukey box plots showing the levels of plasma DHCA and THCA at baseline, study end (after 9 or 21 months of CA treatment) and at follow-up (6–12 months after discontinuation of CA). Only the levels of patients for which follow-up values were available are shown (n = 16). e, f Tukey box plots showing the effect of CA on plasma levels of fibroblast growth factor 19 (FGF19) and 7α-hydroxy-4-cholesten-3-one (C4) after 3, 9, 15 and 21 months of treatment. A control reference range for FGF19 and C4 in children is not available. Two individual C4 data points (patient #2 after 15 months of treatment and patient #15 after 21 months of treatment) were excluded from the analysis, since these measurements were prone to interference due to technical issues. All statistical analyses were performed with a Wilcoxon matched-pairs signed-rank sum test. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001, ns not significant. -2yr = 2 years prior to start of CA treatment
The median FGF19 levels increased significantly upon CA treatment after 3, 9, 15 and 21 months of treatment, whereas the median C4 levels significantly decreased compared to the median baseline levels. This is compatible with suppression of the bile acid synthesis through bile acid-induced upregulation of FGF19 in the terminal ileum (Fig. 1e, f). Also when excluding baseline data from the patients who did not complete 21 months of treatment, the median FGF19 and C4 levels after 15 and 21 months of treatment remained significantly altered compared to baseline (data not shown). No significant differences were observed between the median FGF19 and C4 levels at baseline and 2 years prior to treatment initiation. The individual FGF19 and C4 levels in plasma for each time point are presented in Supplementary Table 1.
Liver tests and elasticity
The median plasma levels of conjugated bilirubin and ALT and AST were normal at baseline and did not significantly change during CA treatment. However, all six patients with elevated conjugated bilirubin levels in plasma at baseline (i.e. patients #16–19, 21 and 22), had a progressive increase of these levels during the treatment course (Fig. 2a, b). The same patients also had liver cirrhosis on ultrasound and/or elastography values of ≥ 15.5 kPa at baseline, indicative for severe liver fibrosis or cirrhosis. No clear trend of plasma ALT and AST in time could be observed in these six patients (red lines in Fig. 2c, d). Additional clinical and biochemical baseline features of the patients with severe liver fibrosis or cirrhosis are presented in Table 2. The median liver elasticity values did not significantly change after 3, 9 or 21 months of CA treatment. The individual ALT, AST, conjugated bilirubin and liver elasticity levels for each time point are presented in Supplementary Table 2.

a Graph showing the individual courses of conjugated bilirubin levels during oral cholic acid (CA) treatment (n = 22). The patients with severe liver fibrosis or cirrhosis based on ultrasound and/or a elastography value of ≥ 15.5 kPa at baseline (n = 6) are depicted in red. The upper control reference range of conjugated bilirubin (7 μmol/L) is indicated by the dotted line. b Box plot (min to max, showing all data points) of the conjugated bilirubin levels of patients with severe liver fibrosis or cirrhosis (n = 6) after 1, 3 and 9 months of CA treatment. The upper control reference range of conjugated bilirubin (7 μmol/L) is indicated by the dotted line. -2yr = 2 years prior to start of CA treatment. c, d Graphs showing the individual courses of alanine transaminase (ALT) and aspartate transaminase (AST) levels during oral CA treatment (n = 22). The patients with liver cirrhosis are depicted in red. The upper control reference ranges of ALT (40 U/L) and AST (45 U/L) are indicated by the dotted lines
Secondary outcome measures
No significant changes were observed in the levels of fat-soluble vitamins A, D or E at any time point (levels normalised for total plasma cholesterol, data not shown). There was a small decrease in PT after 15 months (median of differences 0.3 s, p = 0.049) and 21 months of CA treatment (median of differences 0.4 s, p = 0.020). However, no significant differences were found in aPTT (data not shown). Similar results were found when excluding the baseline data from the patients who did not completed 21 months of treatment. No significant changes were found in plasma total cholesterol levels or albumin levels (data not shown). Also, no significant differences in the median SD scores for weight were observed after 9 months (− 1.05 baseline SD vs. − 1.10 SD, p = 0.71) and after 21 months for the patients who completed the extended treatment phase (− 1.01 baseline SD vs. − 0.95 SD, p = 0.33). The individual weight and median SD score for weight at each time point are presented in Supplementary Table 2.
Fibroblast growth factor 21
Fibroblast growth factor 21 (FGF21) is an endocrine factor produced by the liver, with circulating levels increased under conditions that ‘stress’ the liver (e.g. amino acid deficiency, liver steatosis, alcohol abuse) (Schaap et al. 2013; Cariello and Moschetta 2014; Ye et al. 2014). Since we aimed to identify a biochemical parameter to predict which individual ZSD patient will likely develop a worsening of liver tests upon CA treatment, we divided the patients into subgroups of patients with (n = 6) and without (n = 16) severe liver fibrosis or cirrhosis based on ultrasound and/or elastography values ≥ 15.5 kPa. Significant differences were found in the baseline FGF21 levels, with cirrhotic patients having markedly elevated levels (median 153 vs. 3742 pg/mL, p = 0.0002) (Fig. 3a). FGF21 levels significantly increased after 3, 9, 15 and 21 months of CA treatment (Fig. 3b). The individual courses of FGF21 levels during CA treatment are shown in Fig. 3c.

a Box plot (min to max, showing all data points) of the baseline fibroblast growth factor 21 (FGF21) levels in patients without liver cirrhosis (n = 16, levels in patients #10 and 21 are measured 1 year before treatment initiation because of missing values at baseline) and patients with liver cirrhosis (n = 6). Liver cirrhosis or severe fibrosis was defined as an elastography value of ≥ 15.5 kPa at baseline and/or signs of cirrhosis on ultrasound. Statistical analysis was performed with a Mann–Whitney U-test. b Tukey box plot showing the effect of oral cholic acid (CA) on plasma levels of FGF21 after 3, 9, 15 and 21 months of treatment. A control reference range for FGF21 is not available. Statistical analysis was performed with a Wilcoxon matched-pairs signed-rank sum test. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001. c Graph showing the individual courses of FGF21 levels during CA treatment (n = 22). The patients with severe liver fibrosis or cirrhosis at baseline (n = 6) are depicted in red. -2yr = 2 years prior to start of CA treatment
Discussion
Here, we investigated the effect of 21 months of CA treatment on biochemical parameters and clinically relevant outcome measures in 17 patients with a ZSD. CA induced a significant suppression of bile acid synthesis during the whole treatment phase, as inferred from a decrease in plasma C4 in these patients, and may be explained by the elevation of plasma FGF19 upon CA treatment. The inhibition of bile acid synthesis was accompanied by a decrease in the plasma levels of the toxic C27-bile acid intermediates DHCA and THCA at all time points, with normalisation in some patients. Despite the biochemical effects after 21 months of CA treatment, no effect on liver tests, liver elasticity, fat-soluble vitamin levels or body weight could be observed.
The discrepancy between the effects on bile acid synthesis and lack of effect on clinically relevant outcome measures is remarkable. For some outcome parameters (e.g. liver tests and elasticity), an explanation could be that the treatment phase is too short to observe differences. In addition, a substantial part of the patients already had normal liver tests and liver elasticity at baseline. However, also in individual patients with clearly abnormal liver tests and/or elasticity, no trend towards improvement could be observed. Moreover, the duration of the treatment period does not explain why no noticeable beneficial effect on the (generally low) fat-soluble vitamin levels or body weight could be found. All patients had impaired fat absorption, indicated by their need for fat-soluble vitamin supplementation. Given the theory that C27-bile acid intermediates are less able to form mixed micelles in the intestinal lumen, consequently leading to malabsorption of dietary fat and fat-soluble vitamins (Van Eldere et al. 1987; Stieger et al. 1997), we should have been able to observe potential beneficial effects of CA supplementation on these parameters in the relatively short term. This is supported by the experiences from primary bile acid therapy in other bile acid synthesis disorders (Clayton 1991; Setchell et al. 2003; Haas et al. 2012). A possible explanation for the lack of effect on the above described parameters could be that CA supplementation does not increase the micellar concentration in the intestinal lumen of ZSD patients enough to reach the critical micelle concentration.
Our results raise the question as to whether targeting bile acid metabolism alone is clinically relevant for ZSD patients. It is largely unknown which biochemical abnormalities make the greatest contribution to the different clinical symptoms in ZSD, and it is assumed that it is the combination of abnormalities that is important for the pathophysiological processes. As mentioned, C27-bile acid intermediates were found to be hepatotoxic, especially in their unconjugated form (Ferdinandusse et al. 2009), and their accumulation is, therefore, generally considered to be the major cause of the liver disease in ZSD. However, patients with α-methylacyl-CoA racemase deficiency, a peroxisomal single-enzyme deficiency due to mutations in the AMACR gene, show a comparable accumulation of C27-bile acid intermediates in plasma. Yet, in the majority of cases, no liver disease occurs, but only adult onset of neurological symptoms (e.g. peripheral neuropathy, retinitis pigmentosa and epilepsy) (Ferdinandusse et al. 2000; Haugarvoll et al. 2013). It should, therefore, be considered to investigate the effect of CA treatment on, for instance, the long-term progression of neurological symptoms in ZSD, instead of the liver phenotype alone. We also advise to include coagulation parameters as secondary outcome parameters in future studies studying the effect of CA. Moreover, it cannot be ruled out that the biochemical abnormalities which are not altered by CA treatment (e.g. accumulation of pristanic acid, VLCFAs) play a crucial role in the pathogenesis and progression of liver disease in ZSD. Interestingly, patient #13 (harbouring PEX10 mutations) predominantly shows a neurological phenotype with normal liver tests and elasticity, but has elevated plasma C27-bile acid intermediates in combination with a normal VLCFA profile.
In addition to the 17 patients who were included in the extended 21 months treatment phase, three newly diagnosed patients (#20–22) were included in the initial treatment phase of 9 months. Two of these patients were known to have severe liver fibrosis or cirrhosis based on ultrasound and/or elastography values ≥ 15.5 kPa. This made a total of 6/22 patients with advanced liver disease in the 9 months treatment phase. All these patients showed progressive elevation of conjugated bilirubin levels in plasma during CA treatment, supporting our earlier notion that CA treatment in patients with advanced liver disease is likely to be hepatotoxic.
We aimed to identify biochemical parameters to predict which individual ZSD patients will likely develop a worsening of liver tests upon CA treatment. FGF21, which was suggested to be a potential biomarker for hepatic stress (Cariello and Moschetta 2014; Ye et al. 2014), was significantly higher at baseline in patients with advanced liver disease. Interestingly, there was also a significant increase in the median FGF21 levels upon CA treatment in the total group of ZSD patients, but the meaning of this finding remains to be elucidated.
Recently, another group reported the results of long-term CA treatment in 20 ZSD patients and claimed significant improvement in serum ALT and AST, improved weight percentiles and a significant decrease in serum conjugated bilirubin levels (Heubi et al. 2017). Those patients were treated with the same dosage, but it is unclear how long the individual patients were treated with CA, and follow-up was not structured. Therefore, an important concern in this study is the phenomenon of long-term biochemical normalisation in ZSD patients with a relatively mild phenotype (Berendse et al. 2016a), a bias which cannot be ruled out, since the interval between measurements was not specified. Additional concerns in this study were addressed in a recently published letter to the editor (Klouwer et al. 2017) and stress the need for additional data about the natural course of liver abnormalities in ZSD.
Taken together, the role of CA treatment in the management of ZSD is far from established. Since ZSD patients with advanced liver disease are not deemed eligible for CA treatment, and a substantial fraction of ZSD patients develops no clear liver phenotype without intervention, prospective long-term studies are necessary to define the subgroup of ZSD patients that may benefit from CA treatment. Despite CA being approved in the United States by the Food and Drug Administration (FDA) for the treatment of patients with a ZSD under a ‘rare pediatric disease priority review voucher’ (FDA 2015), we believe that the evidence for possible beneficial effects of CA treatment in ZSD is currently too limited to recommend CA as a therapy in clinical practice. For ZSD patients that are already on CA, it is the responsibility of the treating physician to be aware that clinical benefit of CA therapy in ZSD is not proven and possible side effects in an individual patient should play an important role in the decision whether to discontinue the therapy in a specific patient. In addition, we strongly recommend against the use of CA in ZSD patients with advanced liver disease.
Abbreviations
- aPTT:
-
Partial thromboplastin time
- ALT:
-
Alanine transaminase
- AMC:
-
Academic Medical Center
- AST:
-
Aspartate transaminase
- C4:
-
7α-Hydroxy-4-cholesten-3-one
- CA:
-
Cholic acid
- CYP7A1:
-
Cholesterol 7α-hydroxylase
- DHCA:
-
3α,7α-Dihydroxycholestanoic acid
- FGF19:
-
Fibroblast growth factor 19
- FGF21:
-
Fibroblast growth factor 21
- PT:
-
Prothrombin time
- THCA:
-
3α,7α,12α-Trihydroxycholestanoic acid
- VLCFA:
-
Very long-chain fatty acids
- ZSD:
-
Zellweger spectrum disorder
References
Berendse K, Engelen M, Ferdinandusse S et al (2016a) Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis 39(1):93–106
Berendse K, Klouwer FCC, Koot BGP et al (2016b) Cholic acid therapy in Zellweger spectrum disorders. J Inherit Metab Dis 39:859–868
Bootsma AH, Overmars H, van Rooij A et al (1999) Rapid analysis of conjugated bile acids in plasma using electrospray tandem mass spectrometry: application for selective screening of peroxisomal disorders. J Inherit Metab Dis 22:307–310
Braverman NE, D’Agostino MD, Maclean GE (2013) Peroxisome biogenesis disorders: biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev 17:187–196
Braverman NE, Raymond GV, Rizzo WB et al (2016) Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab 117:313–321
Cariello M, Moschetta A (2014) Fibroblast growth factor 21: a new liver safeguard. Hepatology 60:792–794
Clayton PT (1991) Inborn errors of bile acid metabolism. J Inherit Metab Dis 14:478–496
de Lédinghen V, Le Bail B, Rebouissoux L et al (2007) Liver stiffness measurement in children using FibroScan: feasibility study and comparison with Fibrotest, aspartate transaminase to platelets ratio index, and liver biopsy. J Pediatr Gastroenterol Nutr 45:443–450
FDA (2015) FDA approves Cholbam to treat rare bile acid synthesis disorders
Ferdinandusse S, Houten SM (2006) Peroxisomes and bile acid biosynthesis. Biochim Biophys Acta 1763:1427–1440
Ferdinandusse S, Denis S, Clayton PT et al (2000) Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat Genet 24:188–191
Ferdinandusse S, Denis S, Dacremont G, Wanders RJA (2009) Toxicity of peroxisomal C27-bile acid intermediates. Mol Genet Metab 96:121–128
Haas D, Gan-Schreier H, Langhans C-D et al (2012) Differential diagnosis in patients with suspected bile acid synthesis defects. World J Gastroenterol 18:1067–1076
Haugarvoll K, Johansson S, Tzoulis C et al (2013) MRI characterisation of adult onset alpha-methylacyl-coA racemase deficiency diagnosed by exome sequencing. Orphanet J Rare Dis 8:1
Heubi JE, Bove KE, Setchell KDR (2017) Oral cholic acid is efficacious and well tolerated in patients with bile acid synthesis and Zellweger spectrum disorders. J Pediatr Gastroenterol Nutr 65(3):321–326
Klouwer FCC, Berendse K, Ferdinandusse S, Wanders RJA, Engelen M (2015) Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis 10:151
Klouwer FCC, Braverman NE, Verkade HJ et al (2017) Letter to the editor: oral cholic acid in Zellweger spectrum disorders: a word of caution. J Pediatr Gastroenterol Nutr 66:e57. https://doi.org/10.1097/MPG.0000000000001763
Koot BGP, van der Baan-Slootweg OH, Bohte AE et al (2013) Accuracy of prediction scores and novel biomarkers for predicting nonalcoholic fatty liver disease in obese children. Obesity 21:583–590
Lenícek M, Vecka M, Žížalová K, Vítek L (2016) Comparison of simple extraction procedures in liquid chromatography–mass spectrometry based determination of serum 7α-hydroxy-4-cholesten-3-one, a surrogate marker of bile acid synthesis. J Chromatogr B 1033–1034:317–320
Schaap FG, Kremer AE, Lamers WH, Jansen PL, Gaemers IC (2013) Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie 95:692–699
Schaap FG, Trauner M, Jansen PLM (2014) Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11:55–67
Schreuder TCMA, Marsman HA, Lenicek M et al (2010) The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol 298:G440–G445
Setchell KDR, Bragetti P, Zimmer-Nechemias L et al (1992) Oral bile acid treatment and the patient with Zellweger syndrome. Hepatology 15:198–207
Setchell KDR, Heubi JE, Bove KE et al (2003) Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Gastroenterology 124:217–232
Stieger B, Zhang J, O’Neill B, Sjövall J, Meier PJ (1997) Differential interaction of bile acids from patients with inborn errors of bile acid synthesis with hepatocellular bile acid transporters. FEBS J 244:39–44
Talma H, Schonbeck Y, Bakker B, Hirasing RA, Buuren SV (2010) Groeidiagrammen 2010. Handleiding bij het meten en wegen van kinderen en het invullen van groeidiagrammen
Van Eldere JR, Parmentier GG, Eyssen HJ et al (1987) Bile acids in peroxisomal disorders. Eur J Clin Investig 17:386–390
Wanders RJA, Ferdinandusse S (2012) Peroxisomes, peroxisomal diseases, and the hepatotoxicity induced by peroxisomal metabolites. Curr Drug Metab 13(10):1401–1411
Wanders RJA, Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332
Ye D, Wang Y, Li H et al (2014) Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1α-mediated antioxidant capacity in mice. Hepatology 60:977–989
Acknowledgements
The authors thank the patients and their families for their cooperation. This work was supported in part by grants from Metakids, Hersenstichting (grant F 2012(1)-102), Axel Foundation and Stichting Steun Emma Kinderziekenhuis AMC, the Netherlands. Furthermore, we thank prof. dr. A.K. Groen from the Academic Medical Center in Amsterdam for the helpful discussions.
Funding
This work was supported in part by grants from Metakids, Hersenstichting (grant F 2012(1)-102), Axel Foundation and Stichting Steun Emma Kinderziekenhuis AMC, the Netherlands.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
F. C. C. Klouwer, B. G. P. Koot, K. Berendse, E. M. Kemper, S. Ferdinandusse, K. V. K. Koelfat, M. Lenicek, F. M. Vaz, M. Engelen, P. L. M. Jansen, R. J. A. Wanders, H. R. Waterham, F. G. Schaap and B. T. Poll-The declare that they have no conflict of interest.
Informed consent
Individual written informed consents were obtained from patients and/or the patients’ parents.
Additional information
Communicated by: Robert Steiner
Electronic supplementary material
Below are the links to the electronic supplementary material.
Supplementary Table 1
(DOCX 31 kb)
Supplementary Table 2
(DOCX 31 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Klouwer, F.C.C., Koot, B.G.P., Berendse, K. et al. The cholic acid extension study in Zellweger spectrum disorders: results and implications for therapy. J Inherit Metab Dis (2018). https://doi.org/10.1007/s10545-018-0194-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10545-018-0194-z