
Abstract
Deterioration of oxygen conditions in water below the halocline has been observed in the Baltic Sea. Deoxygenation is linked to the reduced frequency and volume of inflows of highly saline surface water from the North Sea (major Baltic inflows—MBIs) in the second half of the twentieth century and the increased organic matter respiration due to eutrophication. In the present study, the impact of worsening oxygen conditions on pyrite content in the Gdańsk Deep (max. depth of 118 m, southern Baltic Sea) sediments was determined. Geochemical parameters (acid volatile sulfides, pyrite sulfur, reactive iron, organic carbon, sedimentation rate and sediment age) were analyzed in relation to the variation in bottom water oxygen concentration and the occurrence of MBI. The obtained results demonstrate that pyrite content in the study area decreased after 1960. The declining pyrite content coincided with the deterioration of oxygen conditions (concentration < 2 ml l−1) in bottom water. In the same period, reactive iron concentration decreased and organic carbon increased in sediment. In the period 1616–1960, average pyrite accumulation rate was 322 µmol m−2 day−1. In the subsequent years, its average accumulation rate decreased to 210 µmol m−2 day−1. Fluctuations of oxygenation of bottom water in the study area were manifested by highly variable degree of pyritization (36 ± 11%) and particulate organic carbon to pyrite sulfur ratio (2.8–37).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In coastal areas of high productivity, sulfate reduction is the most important pathway of organic matter mineralization in sediment (Jørgensen 1982). One of the products of dissimilatory sulfate reduction is hydrogen sulfide, which may react with reduced iron to form pyrite (Raiswell and Canfield 2012). Pyrite is the iron sulfide phase stable in anoxic environment over a wide range of Eh and pH (Schoonen 2004). Therefore, formation of that mineral provides an important pathway of the removal of iron and sulfur from their cycles in the marine environments (Raiswell and Canfield 2012). Iron is the element that controls the primary production of phytoplankton in the ocean (Martin 1990), and thus affects the sequestration of carbon from the atmosphere (DeVries et al. 2012). As a result, the biogeochemical cycle of Fe can be a key regulator of climate change, which also influences ocean acidification (Martin 1990; Boyd et al. 2007). In this context, it is important to determine factors governing the accumulation of pyrite in sediments of different environments and consequently the removal of Fe from the water.
The process of pyrite formation can be schematically described by the following reactions (Berner 1964, 1974):
Iron monosulfide, being an intermediate product of that process, is transformed into pyrite by oxidation with hydrogen sulfide (Eq. 3) or in the reaction with polysulfide (Wilkin and Barnes 1996; Rickard and Luther 1997). The reaction with hydrogen sulfide is faster than with polysulfide (days vs. years) and dominates in anaerobic sediments (Butler et al. 2004). As it results from reactions 1–3, organic matter, sulfates and Fe oxide-hydroxide (reactive iron, FeR) are essential for the formation of pyrite in sediments. Depending on the environmental conditions, each of these substrates may be a factor limiting the described process. If bottom water is not oxygen-depleted, the H2S necessary for the formation of pyrite is produced during decomposition of organic matter in anaerobic sediment layers. Under such conditions, pyrite is formed after sediment deposition and is then called diagenetic pyrite (Raiswell and Berner 1985). The rate of diagenetic FeS2 formation depends mainly on the sedimentation rate and on the amount of organic matter being deposited (Berner 1982). Consequently, concentrations of organic matter and FeS2 are linearly related (Raiswell and Berner 1985; Álvarez-Iglesias and Rubio 2012). When H2S is present not only in sediment but also in the near-bottom water, pyrite can be formed already in the water column and at the sediment–water interface—it is then called syngenetic pyrite. In this case, FeS2 formation is limited by the availability of FeR. In such environments, a high concentration of pyrite sulfur is often accompanied by high values of Fe and low values of POC in sediment (Raiswell and Berner 1985; Raiswell and Canfield 1998; Canfield et al. 1992; Sternbeck and Sohlenius 1997; Álvarez-Iglesias and Rubio 2012). A potentially limiting role of Fe in the formation of pyrite was discovered by Berner (1970). He introduced the so-called degree of pyritization (DOP) which determines what part of FeR has been transformed into FeS2. For sediment with high content of acid volatile sulfides (AVS), Boesen and Postma (1988) proposed the use of degree of sulfidization (DOS) as an alternative to DOP.
In the Baltic Sea, where the present study was carried out, pyrite is a common mineral, not only in deep water regions, where suboxic and anoxic conditions are dominant for most of the time, but also in coastal regions, where oxygen deficiency occurs seasonally (e.g. Neumann et al. 2005). The Baltic is a shallow, brackish shelf sea with permanent stratification and limited water exchange with the ocean (water residence time is about 25–35 years; Matthäus and Nausch 2003). Surface waters are separated from more saline deep waters by a halocline that usually occurs at a depth of 60–80 m. The concentration of oxygen in water under a halocline depends mainly on the supply of organic matter and on the inflows of more saline and well-oxygenated surface waters from the North Sea during the so-called major Baltic inflows (MBIs) (Mohrholz et al. 2015). These barotropic inflow events are driven by sea level differences between the Kattegat and the Arkona Basin, caused by wind and air pressure forcing (Matthäus 2006; Mohrholz et al. 2015). Denser water from the North Sea flows through a series of shallow sills in the Danish Straits. Before 1970s, MBIs were observed more or less regularly with frequency of 5–7 events per decade (Feistel et al. 2008). Since the mid-1970s, the frequency and volume of inflows have decreased significantly, and only few MBI occurred in subsequent years (Feistel et al. 2008). The long-lasting stagnation of bottom water has become common state in the Baltic (Mohrholz et al. 2015). Long periods of stagnation coincided with progressive eutrophication of the Baltic Sea, the beginning of which dates back to around 1960 (Elmgren 2001). In the last half-century, the area of the sea bottom covered with hypoxic and anoxic waters has increased from around 40,000 to over 60,000 km2 (Carstensen et al. 2014). It is difficult to indicate the extent to which anthropogenic pressure has been responsible for these changes because they may also have been caused by the natural evolution of the Baltic Sea. The pyrite formation in marine environment depends on the concentration of sulfide, the availability and reactivity of iron minerals, and the contact time of both components (Canfield et al. 1992). For this reason, the declining oxygen conditions should be accompanied by a change in the pyrite accumulation in sediments. In order to determine the nature of these changes, the following parameters: pyrite, acid volatile sulfides (AVS), reactive iron (FeR) and particulate organic carbon (POC), were analyzed in dated sediments (cores of approx. 80 cm) collected from one of the Baltic deeps—the Gdańsk Deep. The variability of the obtained results was investigated in relation to changes in the volume of ocean water inflows and oxygen concentration in the bottom water during the last 140 years. Finally, we have drawn conclusions on the general model of changes in pyrite content in the areas with variable oxygen conditions in the near-bottom water.
Materials and methods
Sampling
Research material was collected during two cruises (08–10. 08. 2015 and 07–08. 02. 2016) on board the R/V Baltica. Each time, samples were taken from three stations located in the area of the Gdańsk Deep (Fig. 1). Temperature (T) and salinity (S) in the bottom water were registered using a CTD profiler (Falmouth Scientific Inc.). Concentration of dissolved oxygen (DO) was measured by potentiometric titration (analytical precision ≤ 1.6%) with automatic titrator (SM-Titrino 702, Metrohm) and combined Pt-ring electrode (Winkler method; Grasshoff et al. 1999).

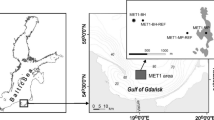
Location of sampling stations in the area of the Gdańsk Deep, southern Baltic Sea (contour lines are depths in meters)
Sediments
From each station, six sediment cores of approx. 80 cm in length were collected: one for analysis of organic carbon (POC), water content (W), loss on ignition (LOI), one for acid volatile sulfides (AVS), total reducible sulfide (TRS) and reactive iron (FeR), three for determination of sedimentation rate and sediment age and one for analysis of hydrogen sulfide and sulfate in pore water. Sediment cores were collected with the Rumohr Lot corer equipped with Plexiglas tubes of 100 cm in length and 7.5 cm in diameter. Cores were sliced into sections and preserved immediately after sampling. The cores for POC, W, LOI were sliced into 1 cm layers. Each layer was placed in a separate polyethylene bag. All sediment samples were frozen at − 21 °C until analysis. The cores for the analysis of AVS, TRS and FeR were sliced into 1 or 2-cm layers (depending on the sampling site), using a PVC ring and a Teflon spatula, in a glove box filled with N2. Samples for determination of AVS and TRS were preserved with zinc acetate immediately after collection and kept frozen (− 21 °C) until analysis. Samples for analysis of FeR were placed in polyethylene bags and frozen at − 21 °C until analysis.
Sedimentation rate and sediment age was determined in 30-cm sediment segments, due to the fact that in deeper sediment layers the activity of 210Pb is below the limit of detection for the applied method. For the purpose of that analysis, cores were sliced into 1-cm layers. Sediment samples from the respective layers of three cores were combined to obtain an analytical sample of suitable weight and to ensure good accuracy of the results.
Pore water
Pore water for hydrogen sulfide and sulfate analysis was retrieved from intact and sealed sediment cores (without any contact with the air), using Rhizon® samplers immediately after sampling. The samplers were inserted through holes drilled in the walls of the liners at 2-cm intervals (from 0 to 40 cm). The first one was placed a few centimeters above the sediment–water interface to collect sample of near-bottom water. Hydrogen sulfide was analyzed with the methylene blue method after sample preservation with zinc acetate (Grasshoff et al. 1999). Precision (RSD) of the hydrogen sulfide determination was 2%. Sulfate concentration was measured using high performance ion chromatography (Methrom 850 Professional IC). Each analysis was performed in triplicate (RSD of measurements was ≤ 3%).
Analytical methods
Water and organic matter content in sediments
Sediment water content (W) was determined by drying samples at 105 °C to the constant mass. Organic matter content was estimated as a gravimetric loss on ignition (LOI) after combustion at 550 °C. Sediment samples for the analysis of organic carbon (POC) were dried at a temperature of 60 °C and homogenized in a porcelain mortar. Samples for POC were acidified with 1 M HCl and combusted in a Perkin Elmer CHNS/O 2400 analyzer (Hedges and Stern 1984). The precision (RSD) of POC assay in sediment samples was ≤ 3%.
Sulfur species
Inorganic S compounds were analyzed as acid volatile sulfides (AVS) and total reduced sulfide (TRS) which represents pyrite sulfur. Concentrations of AVS and TRS were determined in duplicates by sequential extraction method based on the scheme introduced by Zhabina and Volkov (1978). A frozen sediment sample was thawed in a N2-filled glove box, homogenized and divided into subsamples. Approximately 3 g of wet sediment was weighted into reaction flask. The AVS (hydrogen sulfide, organic S complexes, FeS, Fe3S4; Morse and Rickard 2004) were removed from samples under a continuous flow of N2 by extraction with 6 N HCl. To prevent oxidation of sulfide with ferric iron liberated by HCl, the acidification step was carried out in the presence of 1 M ascorbic acid which acts to reduce ferric iron and thus protects the dissolved sulfide (Hsieh et al. 2002). The residue from the AVS leaching was analyzed for TRS using extraction with hot CrCl2 in acidic (12 N HCl) solution under anoxic conditions (Fossing and Jørgensen 1989). In both steps, sulfides produced during distillation were removed in a N2 stream, fixed with 5% Zn acetate and measured with the methylene blue method (Cline 1969). The amounts of AVS and TRS extracted from wet sediment were recalculated on dry weight using the water content in sediment. The precision values of AVS and TRS determination in sediment samples were affected by the natural inhomogeneity of wet sediment samples and were ≤ 3 and ≤ 9% respectively. Similar precision of TRS determination was reported for example for shelf and slope sediments off the coast of Peru (Suits and Arthur 2000).
Total and reactive iron
Reactive iron (FeR) in sediment was determined using 24-h cold 1 M HCl extraction (e.g. Leventhal and Taylor 1990). Sediment samples were freeze-dried and homogenized in agate mortar prior to analysis. About 0.5 g of sediment was weighted into acid-washed HDPE vials and 10 ml of 1 M trace metal grade HCl was added. Samples were shaken for 24 h and then centrifuged (10 min, 3500 revolutions per min). The obtained extract was diluted (1:10) with 0.1 M trace metal grade HNO3 and analyzed by flame absorption atomic spectroscopy (FAAS) with Perkin Elmer spectrometer.
For the determination of total iron (Fetot), about 250–300 mg subsamples of dry sediment were weighed into Teflon digestion vessels. The subsamples were mineralized in a microwave digestion system (Milestone Inc.) with a mixture of trace metal grade concentrated acids: HNO3, HF and HCl v/v 1:2:1. The mineralized subsamples were diluted (1:100) and Fetot concentration was measured by FAAS (Perkin Elmer). The precision of FeR and Fetot assay in sediment samples (RSD) was ≤ 1.5%.
The rate of sedimentation and sediment age
The freeze-dried sediment samples were homogenized prior to analysis. They were placed in plastic containers of identical geometry to that used for calibration. Activity concentrations of 210Pb, 137Cs, 214Bi and 214Pb were analyzed with high resolution gamma spectrometry using a High Purity Germanium (HPGe) detector with a relative efficiency of 40%, and a resolution of 1.8 keV for the peak of 1332 keV of 60Co. The detector was coupled with an 8192-channel computer analyzer (GENIE 2000). The time of measurement was 80,000 s for each sample. 210Pb was determined by gamma emission at 46.5 keV, 226Ra was determined by the emission of its daughter nuclides 214Pb and 214Bi at 352 and 609 keV respectively, and 137Cs was measured via its emission at 661.6 keV. For efficiency calibration, standard solution of gamma emitting isotopes (Code BW/Z-62/27/07, produced at the Institute of Atomic Energy POLATOM, Poland) was used. Standard source was prepared in the same geometry of cylindrical containers with 40 mm diameter as applied for environmental samples.
In the present study, the models based on analysis of the variability in 210Pb activity concentrations along vertical profiles were used to determine the sedimentation rate and age of individual sediment layers (Robbins 1978; Appleby and Oldfield 1992; Appleby 1997; Zajączkowski et al. 2004; Zaborska et al. 2007; Suplinska and Pietrzak-Flis 2008; Mulsow et al. 2009; Diaz-Asencio et al. 2009). The exponential character of changes in radioactive concentrations allowed the use of two models: CRS (Constant Rate of Supply model) and CF:CS (Constant Flux Constant Sedimentation Rate model). The method of their application has been described in detail by Szmytkiewicz and Zalewska (2014). The sediment dating method based on the vertical distribution of 210Pb concentrations has been verified by measuring 137Cs activity changes along the vertical profiles of bottom sediments at all stations. The appropriate events that increased 137Cs concentrations in the Baltic environment were identified in the time period covered by the dating.
To determine the age of individual sediment layers, the CRS model was used. The obtained relationship between depth and age of individual sediment layers was linear and statistically significant (R2 = − 0.99; p = 0.00). The linear equation was used to assign age to sediment layers below the depth covered by actual measurements, i.e. layers below 25 cm. As a result of extrapolation beyond the measurement range, the deepest layers at a depth of 80 cm were found to originate from the period between 1605 (station P1) and 1677 (station P1A).
Pyrite concentration, accumulation rate and degree of pyritization and sulfidization
Pyrite (FeS2) concentration (mg g−1 dw (dry weight)) was calculated from total reducible sulfide (TRS, mg g−1 dw) in sediment:
where \(M_{{FeS_{2} }}\) and \(M_{S}\) is molar mass of pyrite and sulfur.
The pyrite accumulation (A) rate (µmol m−2 day−1) was calculated as:
where d—sediment bulk density (2.6 g cm−3; Alkal 1972), \([FeS_{2} ]\)—pyrite concentration (µmol g−1 dw), ω—linear sedimentation rate (cm day−1) and W—water content (%).
Degree of pyritization (DOP) was calculated according to Berner (1970):
Degree of sulfidization (DOS) was calculated according to Boesen and Postma (1988):
Content of iron in the form of monosulfide (FeFeS) in sediment was calculated based on the concentration of AVS, assuming that this form of sulfur consists mainly of FeS.
Changes in oxygen concentration in bottom water and occurrence of major Baltic inflows
Oxygen profiles for over 8000 locations within the Gdańsk Basin, together with the locations of sampling sites and information on the depths at which the measurements had been taken, were extracted from the ICES Dataset on Ocean Hydrography (ICES 2017). Dataset containing oxygen values in bottom water of the Gdańsk Deep was compiled in ArcGIS 10. From the original dataset, only sites with max. depth > 100 m were selected. For each station, the oxygen values which had been measured at the maximum depth for a given depth profile were selected in the next step. Average monthly values of oxygen were computed for the further analysis.
Information on the occurrence and intensity of major Baltic inflows in years 1880–2007 was taken from the Digital Supplement of the book of Feistel et al. (2008). The measure of the intensity of inflows (QFM96) is based on the total amount of salt, which is transported through the Belt Sea and the Sound into the Baltic Sea during an inflow event (Fischer and Matthäus 1996; Mohrholz et al. 2015).
Statistical methods of data processing and analysis
In order to characterize changes in oxygen concentration in the near-bottom water and the content of pyrite, AVS, POC and FeR, the locally weighted scatterplot smoothing (LOWESS) method was used (Cleveland 1979). It is often applied in environmental studies to smooth lines through scatter plot or time plot (Chandler and Scott 2011). We applied Statistica v.11 software for statistical data processing and analysis.
Results
Conditions in bottom water
Temperature and salinity in the near-bottom water were higher in summer than in winter (Table 1). Higher salinity values measured in summer were a consequence of the strong inflow of waters from the North Sea, which occurred in December 2014, and reached the Gdańsk Deep in January 2015 (Mohrholz et al. 2015; Rak 2016). When the samples were collected, in August 2015, about 7 months following the inflow, the hypoxia was observed in the bottom water of the Gdańsk Deep (oxygen saturation of 11–19%). During the next voyage, in February 2016, concentration of DO in bottom water was similar (Table 1); bottom water salinity, however, was lower than in August 2015 (Table 1). This was a consequence of the arrival of moderate inflow which took place in November 2015 and reached the Baltic Proper in January/February 2016 (Hansson and Andersson 2016).
In any of the research periods we did not record anoxia in the bottom water (about 1 m above sediment). Despite only small differences (max. 2 m) in the depth of the research stations, its influence on the oxygen concentration was reported. During both cruises, the lowest oxygen concentration was measured at the deepest station (P1), and the highest at the shallowest one (P1B) (Table 1).
Hydrogen sulfide and sulfate in pore water
Concentration of hydrogen sulfide in pore water varied from 2 to 2860 µmol l−1 (Fig. 2a). The general trend was increasing concentration with increasing sediment depth. Concentration in sediment layer above 15 cm in August 2015 was lower than in February 2016. In both periods, the concentration rapidly increased in layers below 15–20 cm. Although the difference in depth between stations was only 1–2 m (Table 1), in both seasons the highest concentration of hydrogen sulfide was measured at the deepest station (P1), where oxygen concentration was the lowest (Table 1). Increasing hydrogen sulfide concentration was accompanied by decreasing (from 27 to 0.06 mmol l−1) concentration of sulfate (Fig. 2b). Sulfate concentration in the top 15 cm of sediment in August 2015 was generally higher than in February 2016. This was related to the occurrence of MBI in December 2014.

Pore-water profiles of hydrogen sulfide (a) and sulfate (b) in the Gdańsk Deep in August 2015 and February 2016
Particulate organic carbon, organic matter and water content of sediment
The values of organic carbon (POC), as well as organic matter (expressed as loss on ignition, LOI) and water content (W) in sediments of all three stations were similar (Table 2). Both POC and LOI reached the highest values in the surface layers of sediment. POC ranged from 26.0 to 79.5 mg g−1 dw and LOI ranged from 7.5 to 30.8%. The lowest values of both parameters were noted in a 30–40 cm sediment layer. In deeper layers, the increasing content of organic matter was observed. Sediment water content was from 62.7 to 96.0% and decreased with sediment depth.
Content of pyrite, AVS, total and reactive iron in sediment
The concentration of pyrite in sediments of the study area varied within a wide range from 1.80 to 28.40 mg g−1 dw (Table 2, Fig. 3). In sediments of the deepest station (P1), there was an increase in FeS2 concentration from the surface to about 60 cm below surface (bsf), observed in both seasons. In the case of the other stations, FeS2 concentration in the sediment profile was variable. The maximum values were found between 30 and 50 cm of sediment (Fig. 3).

Profiles of pyrite in sediments of the Gdańsk Deep in August 2015 (P1-VIII, P1A-VIII and P1B-VIII) and February 2016 (P1-II, P1A-II and P1B-II). Solid line represents pyrite concentration smoothed with locally weighted polynomial regression (LOWESS)
The content of reactive iron (FeR) ranged from 4.2 to 15.2 mg g−1 dw, while total iron (Fetot) was between 30.4 and 107.5 mg g−1 dw (Table 2). In the upper 30–40 cm of the sediment, the share of FeR in Fetot was larger (12–31%) and more variable than in deeper sediment layers, where it reached constant values of 8–10%. The concentrations of FeS2 and FeR showed approximately the inverse relationship, except for the surface sediment layer, where in some cases the changes in pyrite and FeR were similar (Fig. 4).

Profiles of acid volatile sulfides (AVS) and reactive iron (FeR) in sediments of the Gdańsk Deep in August 2015 (P1-VIII, P1A-VIII and P1B-VIII) and February 2016 (P1-II, P1A-II and P1B-II). Solid line represents AVS and FeR concentration smoothed with locally weighted polynomial regression (LOWESS)
In the analyzed sediments, the AVS content was much lower than the pyrite content (Figs. 3, 4), and varied from < 0.03 (detection limit) to 1.59 mg g−1 dw (Table 2). The median of FeS2:AVS ratio, which indicates how much Fe occurs in the form of pyrite or in the form of metastable iron sulfides (Berner et al. 1979), was 58. The range of FeS2:AVS was 5–812 with 75% of results > 26. The FeS2:AVS ratio differed depending on the station. The highest values (median = 94, 75% of results in range from 44 to 812) were reported for sediments of the shallowest station (P1B), and the lowest (median = 41, 75% of results in the range of 5–89) for sediments of the deepest station (P1). The accumulation of AVS in sediments reflects oscillations of bottom water oxygen concentration. During periods of improved oxygen conditions, most of the AVS in the surface sediment layer is oxidized (Schippers and Jørgensen 2002). This greatly favors the accumulation of pyrite which is much less susceptible to oxidation (Sternbeck and Sohlenius 1997). As a result, the FeS2:AVS ratios were highest in the area of shallower P1B station, where bottom water oxygen concentration was higher than at other sites (Table 1).
Degree of pyritization
The degree of pyritization (DOP) in the analyzed sediments (Fig. 5) varied over a wide range from 10 to 67% (median = 38.5, mean ± SD = 38.3 ± 13.2). At the deepest station (P1), DOP increased with sediment depth, reaching the value of 59%. At the other stations, DOP varied similarly to FeS2 (Figs. 3, 5). At the P1A station, relatively small DOP values prevailed in the profile (Table 2), with minimum values in the intermediate sediment layers (about 25–40 cm bsf) (Fig. 5). In these layers the highest concentration of AVS (up to 1.59 mg g−1 dw) and FeR was recorded (Fig. 4).

Variability of degree of pyritization (DOP) and sulfidization (DOS) in the sediment profile of the Gdańsk Deep, in August 2015 (P1-VIII, P1A-VIII and P1B-VIII) and February 2016 (P1-II, P1A-II and P1B-II). Solid line represents DOP and DOS smoothed with locally weighted polynomial regression (LOWESS)
The degree of pyritization in the analyzed sediments was approximately equal to the degree of sulfidization (DOS) (Fig. 5). This fact, supported by small concentrations of AVS and high FeS2:AVS ratio, indicates that the dominant combination of reduced Fe and S in the Gdańsk Deep sediments is pyrite.
Sedimentation rate and sediment age
The values of linear accumulation rate (LAR) and mass accumulation rate (MAR) obtained for individual stations were similar (Table 3). The lowest sedimentation rate of 2.1 mm year−1 was determined for station P1B. At stations P1A and P1, linear sedimentation rate values were 2.2 and 2.4 mm year−1 respectively. The mass sedimentation rate was almost identical for the three examined locations (550–560 g m−2 year−1). Based on the obtained sediment dating results, it was calculated that the average age of the cores was 380 years, i.e. the oldest collected sediments were deposited around year 1605.
Long-term changes in bottom water oxygen concentration and occurrence of major Baltic inflows
In the second half of 20th century, the frequency and intensity of major Baltic inflows (MBIs) have decreased (Fig. 6a). Only two very strong inflows (intensity index > 30) were recorded after 1950. The frequency of MBIs in the last 30–40 years has decreased to only one inflow per decade. Long stagnation periods have caused considerable deterioration in oxygenation of bottom water in the Gdańsk Deep (Fig. 6b).

The occurrence and intensity index of Major Baltic Inflows (data from Feistel et al. 2008) in years 1880–2014 (a), and long-term changes of dissolved oxygen concentration in the bottom water of the Gdańsk Deep (b)
Discussion
Content of pyrite in sediments of the Gdańsk Deep—comparison with other regions of the Baltic Sea
In anaerobic sediments of the other Baltic deeps, i.e. Bornholm and Gotland Deep, the formation of pyrite is limited by FeR, and the rate of this process may vary considerably, depending on sedimentation rate, the presence or absence of H2S in the bottom water and salinity (Boesen and Postma 1988). In the Gotland Deep sediments, pyrite concentration ranges from 5 to 80 mg g−1 dw (Boesen and Postma 1988; Sternbeck and Sohlenius 1997), while in the Bornholm Deep—from several up to approx. 38 mg g−1 dw (Boesen and Postma 1988). Similar pyrite content (0–70 mg g−1 dw) is observed in the Holocene sediments of the euxinic Landsort Deep, which is the deepest basin of the Baltic Sea (Hardisty et al. 2016). In contrast, in sediments of the shallow Aarhus Bay, the FeS2 content is from 5 to 14 mg g−1 dw (Holmkvist et al. 2011). However, the concentration of pyrite in shallow water regions is not always lower than in deep water basins. This has been shown, for example, by Neumann et al. (2005), who examined sediments in the Odra estuary, and found the FeS2 content to be 75 mg g−1 dw. In view of these results, the Gdańsk Deep sediments are characterized by relatively low pyrite content (from 2 to 28 mg g−1 dw, on average 10 ± 4 mg g−1 dw), which is the closest to that reported for the Bornholm Deep. As compared to the Gotland Deep (max. depth 249 m) and the Landsort Deep (max. depth 459 m), the Gdańsk (max. depth 118 m) and Bornholm (max. depth 100 m) deeps are significantly shallower, and their bottom waters usually have better oxygen conditions. Anoxia in the bottom water in the Gdańsk and Bornholm Deep is seasonal phenomena (Boesen and Postma 1988; IMGW 2017). For most of the time, bottom waters in the Gdańsk Deep are low oxygen or hypoxic (oxygen concentration < 2 ml l−1). Hydrogen sulfide usually appears from August to October (IMGW 2017). In contrast, the Gotland and Landsort deeps are continuously hypoxic with hydrogen sulfide present in the deepest parts (Boesen and Postma 1988; Zillén et al. 2008). Here pyrite is formed in the anoxic water column (e.g. Boesen and Postma 1988). As a result, concentration of this mineral in the deepest areas of the Baltic is high compared to sediments of the Gdańsk Deep. The average pyrite accumulation rate estimated for sediments of the Gdańsk Deep was 287 µmol m−2 day−1 (SD = 171 µmol m−2 day−1, median 259 µmol m−2 day−1) (Table 4). A pyrite accumulation rate similar to that in the Gdańsk Deep was found for the sediments of the Baltic Sea—North Sea transition area (average value of 330 µmol m−2 day−1, and a median of 240 µmol m−2 day−1) (Jørgensen et al. 1990).
Degree of pyritization and organic carbon to pyrite sulfur ratio
The extent to which Fe in sediments is sulfidized through reaction with sulfides (Eqs. 2–3) may be expressed as a degree of pyritization (DOP). This is a frequently used indicator of euxinic conditions, both for ancient and modern sediments (e.g. Raiswell and Berner 1985; Álvarez-Iglesias and Rubio 2012; Hardisty et al. 2016). Nevertheless, this indicator has some limitations. For example, in ancient sediments the biogenic pyrite formation may proceed long after sedimentation and be unrelated to conditions in the bottom water (Rickard 2012). In turn, in modern sediments, DOP can be controlled by sulfide concentration and exposure time as well as iron mineralogy (Raiswell and Canfield 1996, 2012). Iron minerals have different reactivity towards hydrogen sulfide. There are highly reactive Fe (oxyhydr)oxides (e.g. ferrihydrite, goethite, lepidocrocite and hematite), which react with dissolved sulfide on a timescale of hours and days, and Fe silicates, which are basically unreactive (Canfield 1989; Dos Santos Afonso and Stumm 1992; Raiswell and Canfield 1996, 2012). Some difficulties in comparing the DOP obtained by different researchers also result from the application of various methods for FeR determination. The most commonly used are: extraction with boiling concentrated HCl, citrate-dithionite buffer or cold 1 M HCl (Berner 1970; Canfield 1989; Leventhal and Taylor 1990). All the mentioned methods elute mainly Fe (oxyhydr)oxides, however, the use of boiling HCl results in the removal of significant proportion of silicate-bound Fe from the sediment (Raiswell et al. 1994).
DOP has been calibrated by Raiswell et al. (1988) against a broad range of sediments from depositional environments that were well-constrained from palaeoecological and sedimentological criteria. They found that normal (oxic) marine sediments have values of DOP < 42%. In restricted (poorly oxygenated bottom water) sediment DOP ranges from 45 to 80% and is > 75% in inhospitable bottom conditions, where little or no oxygen is present, and hydrogen sulfide may be continually or periodically present. DOP calculated for sediments of the Gdańsk Deep varied from 15 to 67% and could be included in the range regarded as characteristic of oxic and restricted sediments (Raiswell et al. 1988; Jørgensen et al. 1990; Sternbeck and Sohlenius 1997).Very similar DOP values (8–65%) were reported by Suits and Arthur (2000) for low oxygen but non-euxinic sediments of the Peru margin. Most of the sediments analyzed in the present study (75%) displayed intermediate DOP values (31–67%). This is close to the values obtained by Canfield et al. (1992) in sediments of the FOAM site in Long Island Sound, USA, with highly sulfidic pore waters beneath the oxic bottom. Intermediate DOP values (30–50%) were also reported for euxinic sites of rapid siliciclastic accumulation in the Black Sea (Lyons and Severmann 2006). The authors argued that values close to the upper limit (42%) for oxic sediments (Raiswell et al. 1988), reflect pyrite formation under iron-limited oxic and suboxic conditions.
Similarly to the DOP, the ratio of organic carbon to pyrite sulfur (POC:TRS) in the Gdańsk Deep was highly variable. The ratio of POC to TRS was originally used by Berner and Raiswell (1984) as a bottom water salinity indicator. In normal marine sediment the POC:TRS is 7.5 ± 4.0 (Berner and Raiswell 1983). In the study area, the POC:TRS ranged from low values typical for euxinic environments, through normal marine to high values observed in freshwater sediments. It was from 2.8 to 37 with 75% of values < 12 and median of 8. Since the pyritization process depends on the concentration of sulfide, the reactivity of iron minerals and the contact time of both components (Canfield et al. 1992, 1996), we propose that the variability of DOP and POC:TRS in the Gdańsk Deep is a consequence of variation in redox conditions during sediment deposition. Low DOP and high POC:TRS reflect decreased rates of sulfate reduction in surface sediment and the absence of hydrogen sulfide in bottom water during MBIs. This may also be an effect of reoxidation of solid sulfides in the surface sediment layer during inflow events. Additional explanation for high POC:TRS observed in the Gdańsk Deep is high sedimentation rate and preservation of sedimentary organic matter under low oxygen conditions (Canfield 1994). The availability of dissolved sulfide for the reaction with iron in the study area increases strongly during long-lasting stagnation periods without any MBI. In these periods, hydrogen sulfide is present in bottom water in concentrations up to 200 umol dm−3 (e.g. Łukawska-Matuszewska and Kielczewska 2016). Under conditions of abundant hydrogen sulfide, pyrite can be formed from detrital Fe minerals before sediment deposition (Raiswell and Berner 1985), and this leads to low POC:TRS ratios. However, as already mentioned, most of the time, bottom water in the Gdańsk Deep has low but non-zero oxygen concentration and hydrogen sulfide is present only in pore water. As a consequence, intermediate DOP values typical for restricted marine conditions prevail.
Another frequently used indicator—DOS, is calculated assuming that AVS consists mainly of FeS (Eq. 7). However, it should be emphasized that besides the metastable Fe sulfides, also organic S compounds (particulate and dissolved organic complexes) and dissolved sulfide can be removed from the sediment with HCl (Morse and Rickard 2004). Therefore, AVS should not be identified only as FeS (Rickard 2012). Nevertheless, the Gdańsk Deep sediments contain little AVS (AVS is 2–3 orders of magnitude smaller than FeS2, Table 2). Consequently, DOP and DOS values are very similar (Fig. 5).
Factors limiting the formation of pyrite
In the marine environment, the factor limiting the formation of pyrite is usually the availability of organic matter or Fe (oxyhydr)oxides (e.g. Raiswell and Berner 1985; Sternbeck and Sohlenius 1997). Concentration of sulfates is usually high, and the rate of sulfate reduction in coastal areas is also high enough to provide sufficient amount of hydrogen sulfide (Berner 1970). However, in the brackish environment, such as the Baltic Sea, it may happen that pyrite formation is limited by availability of sulfate for reduction (Boesen and Postma 1988). Moreover, sulfate reduction rate within the surface sediment layer varies seasonally (Aller 1980). Metabolic rate and hydrogen sulfide production decreases during the winter months when temperature and the input of organic matter to the bottom is low (Hardisty et al. 2018).
Hydrogen sulfide
Concentration of sulfate in pore water of the Gdańsk Deep was similar to that measured in other deep water sediments of the Baltic Sea (Carman and Rahm 1997). The downward decrease in sulfate, resulting from dissimilatory microbiological reduction and anaerobic oxidation of methane, was observed for all sampling sites (Fig. 2). Decreasing concentration of this component was accompanied by increasing hydrogen sulfide. In both study periods, concentration of hydrogen sulfide was similar to the values obtained in similar sediment layers in other studies carried out in the Gdańsk Basin (Brodecka et al. 2013; Łukawska-Matuszewska and Kiełczewska 2016; Lukawska-Matuszewska 2016) and Bornholm Basin (Boesen and Postma 1988). It was also considerably higher than the values measured in pore water of the Gotland Deep by Boesen and Postma (1988). The processes such as reaction with highly reactive iron minerals, diffusion to near bottom water and reoxidation potentially decrease sulfide concentration in the surface sediments (Boesen and Postma 1988; Canfield et al. 1992). The oxidation of hydrogen sulfide caused by MBI of 2014 was reflected in pore water profiles, where concentration of this component in the upper 15 cm in August 2015 was considerably lower than in February 2016 (Fig. 2a). As a consequence, pyrite concentration in surface sediment may be to some extent controlled by the availability of sulfide. The fact that pyrite formation may be limited by the availability of hydrogen sulfide is also demonstrated by relatively low DOP and elevated AVS in sediment layers with high content of FeR (20–40 cm) at station P1A (Figs. 4, 5). Concentration of sulfide was probably insufficient to convert AVS to pyrite, which is consistent with two-step mechanism of formation of this mineral as proposed among others by Berner (1964) and Wilkin and Barnes (1996). However, DOP values are similar to DOS in most of the analyzed sediments (Fig. 5), which implies that sulfide controls pyrite formation occasionally and most of sulfidized reactive iron in the study area is present in the form of pyrite.
Hydrogen sulfide concentration increased in deeper (below 20–40 cm) layers of the sediment which was accompanied by decreasing content of reactive Fe (Figs. 2, 4). It has been demonstrated that hydrogen sulfide accumulates in pore water only after complete sulfidization of the most reactive iron oxides (ferrihydrite, goethite, lepidocrocite and hematite) which react with sulfide on time scales of less than 1 year (Canfield 1989; Canfield et al. 1996). For comparison, magnetite reacts with sulfide about 104–106 and Fe-containing silicates about 108 more slowly (Canfield et al. 1992). High pore water concentrations of hydrogen sulfide below 20–40 cm in the Gdańsk Deep reflect low reactivity of iron minerals present in this layer of sediment. The rate of sulfate reduction is greater than the rate of reaction between sulfide and iron and, as a result, hydrogen sulfide builds up in pore water (Fig. 2). Similar situation was observed for example by Canfield et al. (1992) in the Long Island Sound, Connecticut and Boesen and Postma (1988) in the Bornholm Deep, Baltic Sea.
Reactive iron and organic carbon
One way of determining which factor limits pyrite formation is to analyze relations between POC and other parameters, i.e. pyrite sulfur, reactive iron and DOP (Raiswell and Berner 1985). Sediments deposited under normal marine conditions (oxygenated bottom water) show a positive linear relationship between pyrite sulfur and organic carbon described by a straight line passing through the origin (Raiswell and Berner 1985; Álvarez-Iglesias and Rubio 2012). In such environment, pyrite is formed after sediment deposition and the rate of mineral formation is controlled by the sedimentation rate and the amount of organic matter (Raiswell and Berner 1985). On the other hand, the availability of easily reducible iron phases is considered as the limiting factor for the pyrite formation in most of the anoxic/euxinic marine environments (e.g. Berner 1984; Raiswell and Berner 1985; Boesen and Postma 1988). Euxinic sediments can be characterized by (1) uniform pyrite sulfur and DOP with increasing content of POC or (2) positive linear relationship between POC and TRS and FeR (Raiswell and Berner 1985; Lyons and Berner 1992).
In the Gdańsk Deep the POC and pyrite sulfur plot (Fig. 7) shows a TRS variability from 1 to 15 mg g−1 dw over the POC range from 26 to about 80 mg g−1 dw. Also DOP and corresponding FeR are highly variable and not related to the POC variability (Fig. 7). This may suggest that FeR is the limiting factor for pyrite formation in the study area. It has been demonstrated, that reactive Fe availability is the principal factor controlling pyrite formation also in other Baltic basins, e.g. Gotland Deep, Bornholm Deep (Boesen and Postma 1988). On the other hand, most of the data points on the POC-TRS plot (Fig. 7a) lie below the line describing relationship between pyrite sulfur and organic carbon in normal marine sediments. The low TRS accompanied with high POC is typical for environments where pyrite formation is limited by sulfur (Berner and Raiswell 1984). However, our results indicate that sulfur limitation in the Gdańsk Deep may occur only during MBIs as a consequence of reoxidation of hydrogen sulfide and decreasing rate of sulphate reduction (see Sect. 4.3.1).

Relationships between particulate organic carbon (POC) and a pyrite sulfur (TRS) b reactive iron (FeR) and c degree of pyritization (DOP) in the Gdańsk Deep sediments. Solid and dashed lines presented on plot a are mean and range of POC:TRS ratios in normal marine sediment deposited under oxic conditions. The regression line for normal marine sediment was plotted using a mean POC:TRS ratio of 2.8 (Berner 1982; Raiswell and Berner 1986)
Despite the lack of linear correlations between pyrite and POC or FeR, some relationships between these parameters can be identified in the sediment profile (Fig. 8). Interestingly, in the sediment deposited before 1960, the changes in POC and pyrite occur in a similar manner, which may indicate that POC had been a limiting factor for pyrite formation at that time (Fig. 8a). Furthermore, an inverse relationship between FeS2 and FeR is noticed for that period (Fig. 8b). After 1960, the changes of these two parameters are similar, they both decrease (Fig. 8b). This is probably caused by limited pyrite formation, affected by FeR. One of the factors responsible for changes in the pattern of pyrite deposition can be variability of oxygen concentration in bottom waters. Long-term data on oxygen concentration in the bottom areas of the Gdańsk Deep (ICES 2017) show a clear decrease of the values, beginning from the mid-1950s (Fig. 6b). Since then, only two very strong MBIs have been recorded (Fig. 6a). In addition, since the mid-1970s, the frequency of lower intensity inflows has also been reduced (Fig. 6a). Another reason for the decrease in oxygen concentration in the bottom water of the Gdańsk Deep over the last several decades is eutrophication. The increased inputs of nutrients from land has increased phytoplankton production and oxygen consumption through enhanced respiration of organic material (Carstensen et al. 2014). Thus, the anthropogenic pressure may be considered to have an impact on geochemical factors which control and limit pyrite formation in marine sediments. However, more work is needed to assess the relative importance of physical processes and eutrophication in this process. On the whole, the worsening oxygen conditions in the study area are reflected in the composition of sediment deposited after 1960, i.e. lower FeR content (Fig. 8). Before 1960, pyrite had been accumulated in sediments with the average rate of 322 μmol m−2 day−1 (Table 4). In subsequent years, when the long-lasting stagnation of bottom water became usual state in the Baltic, pyrite accumulation decreased and was on average 210 μmol m−2 day−1 (Table 4). The investigated sediments are relatively young, therefore we cannot exclude the possibility that lower pyrite accumulation rate in sediment deposited after 1960 is to some extent related to the incomplete Fe pyritization. However, the (oxyhydr)oxides are the main ferric iron minerals reacting with dissolved sulfide on early diagenetic time scales in coastal areas (Raiswell and Canfield 1996, 2012). Considering the reaction time of most sedimentary Fe minerals with dissolved sulfide (see Sect. 4.3.1), it can be expected that the most reactive phases have already reacted. In addition, the concentration of AVS in this sediment layer is small and its complete conversion into pyrite, would increase the accumulation of this component by only 6 ± 5 μmol m−2 day−1.

Changes in the average content of a organic carbon (POC) and pyrite, b reactive iron (FeR) and pyrite and c POC and FeR, with the age of sediments in the Gdańsk Deep. For the purpose of these comparisons, only layers deposited after 1700 were used. The average values of the analyzed parameters were calculated based on results obtained for particular sediment layers deposited in the same year at all examined stations. Gray area indicates pyrite accumulation rate in the Gdańsk Deep. On the right panel, the variability of analyzed parameters (POC, pyrite and FeR) in sediment smoothed with locally weighted polynomial regression (LOWESS) together with the occurrence and intensity of major Baltic inflows (MBI; bars –intensity of inflows; line—LOWESS) is presented. For years in which more than one inflow occurred the volume of all events were summed
After 1960, the concentration of POC in sediments has increased significantly and concentration of FeS2 and FeR decreased (Fig. 8). Such relationship can be brought about by FeR consumption during organic matter mineralization (Froelich et al. 1979; Raiswell and Canfield 2012). In addition, with limited oxygen resources, the oxidation of Fe2+, which is a source of FeR, is less intensive. The impact of improvement of bottom water oxygenation following MBIs on the FeR formation has been demonstrated for example in the Gotland Deep where large amounts of iron and manganese precipitated and dissipated from the water column (Yakushev et al. 2011). The oxidation of Fe2+ was also evident in the bottom water of the Gdańsk Deep as a suspended particulate matter collected in August 2015 (7 months after MBI) had light orange colour as an effect of Fe oxy(hydroxides) precipitation (Fig. 1S, suppl. material). As a consequence, positive relationship between inflows and pyrite was observed (Fig. 8b). The positive dependence between FeS2 and the inflows in the period from around 1920 until 1980s potentially also results from the fact that some oxidation of the FeS surface accelerates pyritization and is required for pyrite formation (Wilkin and Barnes 1996). The reaction of iron sulfide and hydrogen sulfide (Eq. 2) can be inhibited if there is no oxidation of the FeS reactant (Wilkin and Barnes 1996; Butler and Rickard 2000). In periods when oxygen conditions in the bottom water improve, the FeS partial oxidation may occur in the topmost sediment layer (Wilkin and Barnes 1996):
This increases the amount of FeR available for the reaction with hydrogen sulfide:
The resulting Fe2+ can be precipitated as FeS or directly as FeS2. The reactions take place as long as fresh FeS surfaces continue to be exposed to weakly oxidizing conditions. The amount of oxygen available at the bottom of the Gdańsk Deep is insufficient to completely oxidize H2S released from sediments (Łukawska-Matuszewska and Graca 2018) as a result of sulfate reduction occurring during mineralization of organic matter or during methane oxidation. Therefore, it is likely that the discussed mechanism has a significant impact on FeS2 formation in the study area. It should be noted, however, that before about 1920, the inflows and FeS2 concentrations were approximately inversely correlated (Fig. 8b). Probably the proportions between oxygen concentration in bottom waters and the amount of organic matter reaching the bottom were such that the improvement of oxygen conditions after the inflows significantly limited reduction of sulfates and, consequently, the formation of iron sulfides.
To summarize, relationships between FeS2, POC and FeR obtained in the present study indicate that before 1960 the formation of pyrite had been more often limited by POC. The importance of reactive iron as a limiting factor increased after 1960 when deterioration of oxygen conditions occurred. The decrease in oxygen concentration around 1950 results mainly from the weakened ventilation of bottom water. In the subsequent years, a further decline in oxygen conditions resulting from eutrophication has occurred in the study area. The excessive nutrient input from land has caused an increase in sedimentation of organic material, leading to the imbalance between oxygen supply from inflows and oxygen consumption in mineralization processes. The deterioration of oxygen conditions have lowered the availability of FeR, which in turn became the limiting factor for pyrite formation.
Conclusions
The findings from the present study demonstrate that fluctuations in the bottom water oxygenation are accompanied by a change in pyrite accumulation in sediments of the Gdańsk Deep. Temporary aerobic conditions following the inflows of highly saline surface water from the North Sea (MBIs) promote acid volatile sulfide oxidation in the surface layer of sediment, which in turn greatly favors the accumulation of pyrite in relation to that of metastable iron sulfides. The improvement in oxygen conditions causes the oxidation of hydrogen sulfide, which becomes the main factor limiting pyrite formation during MBIs. The changes in oxygen conditions in bottom water are reflected in highly variable degree of pyritization and organic carbon to pyrite sulfur ratio in sediments of the study area.
The variability of pyrite, organic carbon and reactive iron content obtained for the study area indicate that, during the period from ca. 1700 to 1960, pyrite formation was limited mainly by the availability of organic carbon. After 1960, the importance of reactive iron as a limiting factor increased. We propose that, the observed alteration of the factor limiting pyrite formation, was caused by expansion of bottom water anoxia and hypoxia due to poor ventilation (i.e. less frequent MBIs) and eutrophication in the study area. The average pyrite accumulation rate in sediments of the Gdańsk Deep in the period of the long-lasting stagnation of bottom water decreased by 35% compared to previous years. Similar effect of prolonged stagnation periods can be expected for other Baltic deeps. However, it does not imply that low activity of inflows reduces pyrite retention in the water body. This is due to the fact that limited ventilation of the Baltic deep water favors the expansion of anoxic bottom where pyrite formation may take place. To explain the discussed processes in detail, further studies are needed, in particular the ones with the use of geochemical modeling.
References
Alkal T (1972) The relationship between the physical properties of underwater sediments that affect bottom reflections. Mar Geol 13:251–266
Aller RC (1980) Diagenetic processes near the sediment-water interface of Long Island sound. I.: decomposition and nutrient element geochemistry (S, N, P). Adv Geophys 22:237–350
Álvarez-Iglesias P, Rubio B (2012) Early diagenesis of organic-matter-rich sediments in a ría environment: organic matter sources, pyrites morphology and limitation of pyritization at depth. Est Coast Shelf Sci 100:113–123
Appleby PG (1997) Dating of sediments by 210Pb: problems and solutions. In: Proceedings of a seminar dating of sediments and determination of sedimentation rate, Helsinki, Finland. STUK A 145:7.
Appleby PG, Oldfield F (1992) Application of 210Pb to sedimentation studies. In: Ivanovich M, Harmon RS (eds) Uranium-series disequilibrium: application to earth, marine, and environmental sciences. Clarendon Press, Oxford, pp 731–778
Berner RA (1964) An idealized model of dissolved sulfate distribution in recent sediments. Geochim Cosmochim Acta 28(9):1497–1503
Berner RA (1970) Sedimentary pyrite formation. Am J Sci 268:1–23
Berner RA (1974) Kinetic models for the early diagenesis of nitrogen, sulfur, phosphorus and silicon in anoxic marine sediments. In: Goldberg ED (ed) The sea, vol 5. Wiley-Interscience, New York, pp 427–450
Berner RA (1982) Burial of organic carbon and pyrite sulfur in the modern ocean: its geochemical and environmental significance. Am J Sci 282:451–478
Berner RA (1984) Sedimentary pyrite formation; an update. Geochim Cosmochim Acta 48:605–615
Berner RA, Raiswell R (1983) Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time: a new theory. Geochim Cosmochim Acta 47:855–862
Berner RA, Raiswell R (1984) C/S method for distinguishing freshwater from marine sedimentary rocks. Geology 12:365–368
Berner RA, Baldwin T, Holdren GR (1979) Authigenic iron sulfides as paleosalinity indicators. J Sediment Res 49(4):1345–1350
Boesen C, Postma D (1988) Pyrite formation in anoxic environments of the Baltic. Am J Sci 288:575–603
Boyd PW, Jickells T, Law CS, Blain S, Boyle EA, Buesseler KO, Coale KH, Cullen JJ, de Baar HJW, Follows M, Harvey M, Lancelot C, Levasseur M, Owens NPJ, Pollard R, Rivkin RB, Sarmiento J, Schoemann V, Smetacek V, Takeda S, Tsuda A, Turner S, Watson AJ (2007) Mesoscale iron enrichment experiments 1993–2005: synthesis and future directions. Science 315:612–617
Brodecka A, Majewski P, Bolałek J, Klusek Z (2013) Geochemical and acoustic evidence for the occurrence of methane in sediments of the Polish sector of the southern Baltic Sea. Oceanologia 55:951–978
Butler IB, Rickard D (2000) Framboidal pyrite formation via the oxidation of iron(II) monosulfide by hydrogen sulphide. Geochim Cosmochim Acta 64:2665–2672
Butler IB, Böttcher ME, Rickard D, Oldroyd A (2004) Sulfur isotope partitioning during experimental formation of pyrite via the polysulfide and hydrogen sulfide pathways: implications for the interpretation of sedimentary and hydrothermal pyrite isotope records. Earth Planet Sci Lett 228:495–509
Canfield DE (1989) Reactive iron in marine sediments. Geochim Cosmochim Acta 53:619–632
Canfield DE (1994) Factors influencing organic carbon preservation in marine sediments. Chem Geol 114:315–329
Canfield DE, Raiswell R, Bottrell SH (1992) The reactivity of sedimentary iron minerals toward sulfide. Am J Sci 292:659–683
Canfield DE, Lyons TW, Raiswell R (1996) A model for iron deposition to euxinic Black Sea sediments. Am J Sci 296:818–834
Carman R, Rahm L (1997) Early diagenesis and chemical characteristics of interstitial water and sediments in the deep deposition bottoms of the Baltic proper. J Sea Res 37:24–47
Carstensen J, Andersen JH, Gustafsson BG, Conley DJ (2014) Deoxygenation of the Baltic Sea during the last century. PNAS 111:5628–5633
Chandler R, Scott M (2011) Statistical methods for trend detection and analysis in the environmental sciences. Wiley, Chichester
Cleveland WS (1979) Robust locally weighted regression and smoothing scatter-plots. J Am Stat Assoc 74:829–836
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
DeVries T, Primeau F, Deutsch C (2012) The sequestration efficiency of the biological pump. Geophys Res Lett 39:L13601
Diaz-Asencio M, Alonso-Hernandez CM, Bolanos-Alvarez Y, Gomez-Batista M, Pinto V, Morabito R, Hernandez-Albernas JI, Eriksson M, Sanchez-Cabeza JA (2009) One century sedimentary record of Hg and Pb pollution in the Sagua estuary (Cuba) derived from 210Pb and 137Cs chronology. Mar Pollut Bull 59:108–115
Dos Santos Afonso M, Stumm W (1992) Reductive dissolution of iron(III) (hydr)oxides by hydrogen sulfide. Langmuir 8:1671–1675
Elmgren R (2001) Understanding human impact on the Baltic. Ecosystem: changing views in recent decades. AMBIO 30:222–231
Feistel R, Nausch G, Wasmund N (eds) (2008) State and evolution of the Baltic Sea 1952–2005. A detailed 50-year survey of meteorology and climate, physics, chemistry, biology and marine environment. Wiley, Hoboken. ISBN 978-0-471-97968-5
Fischer H, Matthäus W (1996) The importance of the Drogden Sill in the Sound for major Baltic inflows. J Mar Syst 9:137–157
Fossing H, Jørgensen BB (1989) Measurement of bacterial sulfate reduction in sediments: evaluation of single-step chromium reduction method. Biogeochemistry 8:205–222
Froelich P, Klinkhammer G, Bender M, Luedtke N, Heath GR, Cullen D, Dauphin P, Hammond D (1979) Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Cosmochim Cosmochim Acta 43:1075–1090
Grasshoff K, Kremling K, Ehrhardt M (1999) Methods of seawater analysis 3., completely rev. and extended ed. Wiley-VCH, Weinheim, p 600
Hansson M, Andersson L (2016) Oxygen survey in the Baltic Sea 2016—extent of anoxia and hypoxia, 1960–2016. SMHI Report Oceanography No. 58
Hardisty DS, Riedinger N, Planavsky NJ, Asael D, Andrén T, Jørgensen BB, Lyons T (2016) A Holocene history of dynamic water column redox conditions in the Landsort Deep, Baltic Sea. AJS 316:713–745
Hardisty DS, Lyons TW, Riedinger N, Isson TT, Owens JD, Aller RC, Rye DM, Planavsky NJ, Reinhard CT, Gill BC, Masterson AL, Asael D, Johnston DT (2018) An evaluation of sedimentary molybdenum and iron as proxies for pore fluid paleoredox conditions. Am J Sci 318:527–556
Hedges I, Stern J (1984) Carbon and nitrogen determinations of carbonate containing solids. Limnol Oceanogr 29:657–663
Holmkvist L, Ferdelman TG, Jörgensen BB (2011) A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim Cosmochim Acta 75:3581–3599
Hsieh Y-P, Chung S-W, Tsau Y-J, Sue C-T (2002) Analysis of sulfides in the presence of ferric minerals by diffusion methods. Chem Geol 182:195–201
ICES (2017) ICES Dataset on Ocean Hydrography. The International Council for the Exploration of the Sea, Copenhagen. 2014. https://ocean.ices.dk/HydChem/HydChem.aspx?plot=yes. Accessed 29 Sept 2017
IMGW (2017) Monitoring data from The Institute of Meteorology and Water Management–National Research Institute. Extracted. http://baltyk.pogodynka.pl//index.php?page=2&subpage=5&data=3. Accessed 02 Nov 2017
Jørgensen BB (1982) Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature 296:643–645
Jørgensen BB, Bang M, Blackburn TH (1990) Anaerobic mineralization in marine sediments from the Baltic Sea-North Sea transition. Mar Ecol Prog Ser 59:39–54. https://doi.org/10.3354/meps059039
Leventhal J, Taylor C (1990) Comparison of methods used to determine degree of pyritization. Geochim Cosmochim Acta 54:2621–2625
Lukawska-Matuszewska K (2016) Contribution of non-carbonate inorganic and organic alkalinity to total measured alkalinity in pore waters in marine sediments (Gulf of Gdansk, S-E Baltic Sea). Mar Chem 186:211–220
Łukawska-Matuszewska K, Graca B (2018) Pore water alkalinity below the permanent halocline in the Gdańsk Deep (Baltic Sea)—concentration variability and benthic fluxes. Mar Chem 204:49–61
Łukawska-Matuszewska K, Kiełczewska J (2016) Effects of near-bottom water oxygen concentration on biogeochemical cycling of C, N and S in sediments of the Gulf of Gdansk (southern Baltic). Cont Shelf Res 117:30–42
Lyons TW, Berner RA (1992) Carbon-sulfur-iron systematics of the uppermost deep-water sediments of the Black Sea. Chem Geol 99:1–27
Lyons TW, Severmann S (2006) A critical look at iron paleoredox proxies: New insights from modern euxinic marine basins. Geochim Cosmochim Acta 70:5698–5722
Martin J (1990) Glacial-interglacial CO2 change: the iron hypothesis. Paleoceanography 5:1–13
Matthäus W (2006) The history of investigation of salt water inflows into the Baltic Sea—from the early beginning to recent results. Mar Sci Rep 65:p73
Matthäus W, Nausch G (2003) Hydrographic-hydrochemical variability in the Baltic Sea during the 1990s in relation to changes during the 20th century, ICES. Mar Sci Symp 219:132–143
Mohrholz V, Naumann M, Nausch G, Krüger S, Gräwe U (2015) Fresh oxygen for the Baltic Sea—an exceptional saline inflow after a decade of stagnation. J Mar Syst 148:152–166
Morse JW, Rickard D (2004) Chemical dynamics of sedimentary acid volatile sulfide. Environ Sci Technol 38:131A+136A
Mulsow S, Piovano E, Cordoba F (2009) Recent aquatic ecosystem response to environmental events revealed from 210Pb sediment profiles. Mar Poll Bull 59:175–181
Neumann T, Rausch N, Leipe T, Dellwig O, Berner Z, Böttcher M (2005) Intense pyrite formation under low-sulfate conditions in the Achterwasser lagoon, SW Baltic Sea. Geochim Cosmochim Acta 69:3619–3630
Raiswell R, Berner RA (1985) Pyrite formation in euxinic and semi-euxinic sediments. Am J Sci 285:710–724
Raiswell R, Berner RA (1986) Pyrite and organic matter in Phanerozoic normal marine shales. Geochim Cosmochim Acta 50:1967–1976
Raiswell R, Canfield DE (1996) Rates of reaction between silicate iron and dissolved sulfide in Peru Margin sediments. Geochim Cosmochim Acta 60:2777–2787
Raiswell R, Canfield DE (1998) Sources of iron for pyrite formation in marine sediments. Am J Sci 298:219–245
Raiswell R, Canfield DE (2012) The iron biogeochemical cycle. Past and present. Geochemical perspectives 1. European Association of Geochemistry. Retrieved June 6, 2016, http://www.geochemicalperspectives.org/online/v1n1. https://doi.org/10.7185/geochempersp.1.1
Raiswell R, Buckley F, Berner RA, Anderson TF (1988) Degree of pyritization of iron as a paleoenvironmental indicator of bottom-water oxygenation. J Sediment Petrol 58:812–819
Raiswell R, Canfield DE, Berner RA (1994) A comparison of iron extraction methods for the determination of degree of pyritisation and the recognition of iron-limited pyrite formation. Chem Geol 111:101–110
Rak D (2016) The inflow in the Baltic Proper as recorded in January–February 2015. Oceanologia 58:241–247
Rickard D (2012) Sulfidic sediments and sedimentary rocks. Developments in Sedimentology. Elsevier Science, Amsterdam. ISBN 9780080931838
Rickard D, Luther GW (1997) Kinetics of pyrite formation by the H2S oxidation of iron(II) monosulfide in aqueous solutions between 25 and 125 degrees C: the mechanism. Geochim Cosmochim Acta 61:135–147
Robbins JA (1978) Geochemical and geophysical applications of radioactive lead. In: Nriagu JO (ed) Biogeochemistry of lead in the environment. Elsevier, Amsterdam, pp 285–393
Schippers A, Jørgensen BB (2002) Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments. Geochim Cosmochim Acta 66:85–92
Schoonen MAA (2004) Mechanisms of sedimentary pyrite formation. In: Amend JP, Edwards KJ, Lyons TW (eds) Sulfur biogeochemistry—past and present: Boulder, Colorado, Geological Society of America Special Paper 379, pp 117–134
Sternbeck J, Sohlenius G (1997) Authigenic sulfide and carbonate mineral formation in Holocene sediments of the Baltic Sea. Chem Geol 135:55–73
Suits NS, Arthur MA (2000) Sulfur diagenesis and partitioning in Holocene Peru shelf and upper slope sediments. Chem Geol 163:219–234
Suplińska MM, Pietrzak-Flis Z (2008) Sedimentation rate and dating of bottom sediments in the Southern Baltic Sea region. Nukleonika 53:1–7
Szmytkiewicz A, Zalewska T (2014) Sediment deposition and accumulation rates determined by sediment trap and 210Pb isotope methods in the Outer Puck Bay (Baltic Sea). Oceanologia 56:85–106
Wilkin RT, Barnes HL (1996) Pyrite formation by reaction of iron monosulfides with dissolved inorganic and organic sulfur species. Geochim Cosmochim Acta 60:4167–4179
Yakushev EV, Kuznetsov IS, Podymov OI, Burchard H, Neumann T, Pollehne F (2011) Modeling the influence of oxygenated inflows on the biogeochemical structure of the Gotland Sea, central Baltic Sea: changes in the distribution of manganese. Comput Geosci 37:398–409
Zaborska A, Carroll J, Papucci C, Pempkowiak J (2007) Intercomparison of alpha and gamma spectrometry techniques used in 210Pb geochronology. J Environ Radioact 93:38–50
Zajączkowski M, Szczuciński W, Bojanowski R (2004) Recent changes in sediment accumulation ratek in Adventfjorden, Svalbard. Oceanologia 46:217–231
Zhabina NN, Volkov II (1978) A method of determination of various sulfur compounds in sea sediments and rocks. In: Krumbein WE (ed) Environmental biogeochemistry and geomicrobiology. Ann Arbor Science Publishers, Michigan, pp 735–745
Zillén L, Conley DJ, Andrén T, Andrén E, Björck S (2008) Past occurrences of hypoxia in the Baltic Sea and the role of climate variability, environmental change and human impact. Earth Sci Rev 91:77–92
Acknowledgements
This study was financed by the Polish National Science Centre (Project No. UMO-2013/11/B/ST10/00322).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Responsible Editor: Leila J. Hamdan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Łukawska-Matuszewska, K., Graca, B., Brocławik, O. et al. The impact of declining oxygen conditions on pyrite accumulation in shelf sediments (Baltic Sea). Biogeochemistry 142, 209–230 (2019). https://doi.org/10.1007/s10533-018-0530-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-018-0530-2