
Abstract
Two lineages of brushtail possums (Trichosurus vulpecula) were historically introduced to Aotearoa New Zealand, and these two subspecies have different phenotypic forms. Despite over 100 years of potential interbreeding, they appear to retain morphological differences, which may indicate reproductive isolation. We examined this using population samples from a confined landscape and scored each specimen for phenotype using a number of fur colour traits. This resulted in a bimodal trait distribution expected for segregated grey and black lineages. We also sought evidence for genetic partitioning based on spatial and temporal effects. Genetic structure and rates of genetic mixing were determined using seven neutral, species-specific nuclear microsatellite markers and mitochondrial DNA control region sequence. Genotype analyses indicated high levels of variation and mtDNA sequences formed two major haplogroups. Pairwise tests for population differentiation of these markers found no evidence of subdivision, indicating that these brushtail possums behave as a single randomly mating unit. Despite maintenance of two main colour phenotypes with relatively few intermediates, previous inference of assortative mating and anecdotes of distinct races, our data indicate that New Zealand brushtail possums can freely interbreed, and that in some locations they have formed completely mixed populations where neutral genetic markers are unrelated to phenotype. This has implications for effective pest management towards eradication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In Aotearoa New Zealand, deliberate introduction of plants and animals, mostly during European colonisation (Thomson 1922), has resulted in extinctions and population decline across the native biota (Paulay 1994; Spatz et al. 2017; Wodzicki and Wright 1984; Woinarski et al. 2015). Today, one of the most abundant mammal pest species in New Zealand is the Australian brushtail possum (Trichosurus vulpecula), which is both a voracious folivore and an efficient arboreal predator of small vertebrates and invertebrates (Cowan 2005; Tompkins 2018). Initially introduced to establish a fur industry (Pracy 1974), this species is now recognised as a crucial challenge to conservation of native biosystems and as a vector of bovine tuberculosis- a major issue for the agriculture economy (Caley et al. 1999).
Brushtail possums in Aotearoa came from several source populations in Australia that are recognised as having distinct phenotypic traits (How and Kerle 1995). Successful establishment followed by deliberate regional translocation resulted in a high level of genetic and phenotypic polymorphism across the country (Taylor et al. 2004; Sarre et al. 2014). This diversity is the obverse of the situation where natural range expansion is usually associated with low genetic diversity, leading to the so-called genetic paradox of invasion (Estoup et al. 2016). The success of some biological invasions has been attributed to advantages conferred by high genetic diversity resulting from intraspecific hybridisation (Benvenuto et al. 2012; Rius and Darling 2014), and this might be relevant given the origin of brushtail possums in New Zealand. The scale of genetic diversity and the pattern of gene flow that establishes within an invaded territory can significantly affect the adaptive potential of populations just as it does in natural populations (Slatkin 1987). In addition to enhancing the likelihood of successful invasion of novel habitats by an introduced species, genetic heterogeneity resulting from lineage mixing is expected to present challenges to pest control measures (Adams et al. 2014; Prior et al. 2018).
In Aotearoa, brushtail possums have no natural predators and reach high density in native habitats as well as anthropogenic environments (Efford 2000; Rouco et al. 2013). Naïve native prey and abundant habitat with vegetation palatable to mammalian folivores have enabled this possum species to occupy most of the landscape except the highest elevations (Clout 2000). In response, population management involves the use of aerial broadcast of 1080 (sodium fluoroacetate) toxin (Atkinson et al. 1995; Eason et al. 2011), other synthetic poisons (Goldson et al. 2015; Parkes and Murphy 2003), trapping and hunting. Pest management and harvesting usually targets high density populations and so contributes to spatial and temporal heterogeneity in pest abundance, which is likely to influence patterns and rates of gene flow. In particular, relatively large-scale aerial application of 1080 with knockdown rates of ~ 90%, could increase the degree of mixing as possums in adjacent habitat disperse into the control zone. A better understanding of density dependent dispersal and associated factors is necessary for efficient control leading to pest eradication (Sullivan et al. 2017). However, reproductive behaviour of the pest could also be influential, especially where cryptic lineages exist within a pest taxon. If discrete reproductive units with different ecology exist in the same landscape, management requirements will need to be applicable to each entity (Bastos et al. 2011; Jarić et al. 2019; Perry et al. 2018).
Reproductive segregation of possum subspecies introduced to different parts of Aotearoa could yield differing pest characteristics, and persistence of distinct lineages could mean different management strategies are required. The brushtail possum population in New Zealand is not genetically homogenous, and despite their short history and rapid population expansion, they are spatially structured (Sarre et al. 2014; Taylor et al. 2004; Triggs and Green 1989) (Fig. 1). At least part of this spatial structure correlates with possum fur colour variation (Sarre et al. 2014; Campbell et al. 2021). In their native range six subspecies of Trichosurus vulpecula are recognised with distinct geographic ranges and coat colour (How and Kerle 1995). Analysis of size, fur colour, skull morphology, allozymes and karyotypes have revealed few diagnostic features among Australian populations but confirmed the status of regionally distinct subspecies within their native range (Kerle et al. 1991). Possums introduced to New Zealand represent two of these subspecies; Trichosurus vulpecula vulpecula from south-eastern Australia and Trichosurus vulpecula fuliginosus from Tasmania (Pracy 1974). In the east of New Zealand’s North Island, possums in adjacent areas retain founder characteristics that result from separate introductions of the subspecies, suggesting pre and/or postzygotic isolation (Sarre et al. 2014). If this partitioning among subspecies applies throughout Aotearoa, efforts to model outcomes of pest management, population recovery and migration in a metapopulation framework will need to incorporate dynamics of this tension before effective control can be achieved (Engler et al. 2019; Quilodrán et al. 2018; Ranke et al. 2020).

Nationwide population genetic structure of invasive brushtail possums (Trichosurus vulpecula) in Aotearoa New Zealand inferred using MCMC Bayesian clustering of variation at five microsatellite loci among 799 possums sampled between 1989 and 1997 (data from Taylor et al. 2004)
We sought to determine the level of reproductive isolation between the two Australian brushtail possum subspecies in Aotearoa within a naturally constrained habitat space. We looked for spatial and temporal effects on genetic structure within our sample, and then looked for genetic structure concordant with fur colour phenotype. The presence of possums with distinct fur colour at the study site and recording colour traits for each individual sampled, allowed us to examine the possibility of non-random mating with mate choice linked to fur colour (Sarre et al. 2014). If brushtail possums in this region are randomly mating without strong fitness disadvantage in resulting hybrids, then we would expect to detect no genetic structure within our sample. However, should there be a mating bias (or hybrid disadvantage), we predicted significant deviations from Hardy–Weinberg proportions as well as genetic differentiation concordant with phenotype.
Methods
Location
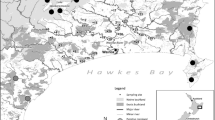
Kenepuru Peninsula is an area of nearly 400 km2 connected by a narrow isthmus in the Marlborough Sounds of northern South Island, New Zealand. The region is characterised by drowned valleys resulting from tectonic subsidence (Trewick and Bland 2012) and the peninsula is largely separated from the mainland with low potential for terrestrial dispersal. Much (~ 60%) of the original forest of Kenepuru Peninsula was removed for pastoral farming by the early twentieth century, but natural regeneration has since formed a mosaic of pasture (~ 20%), scrub and forest.
We used a sample of 132 individual possums from the Kenepuru Peninsula collected during three different time periods (Table 1; Fig. 2). Each possum was photographed and sexed, with date and location recorded. Tissue samples, such as ear clips or muscle biopsies, were taken from fresh possum corpses by landowners and pest controllers and stored in 95% ethanol. For each sample we sequenced a region of the mitochondrial D-loop and determined allelic variation at 7 microsatellite loci.

The Kenepuru Peninsula, Marlborough Sounds, Aotearoa New Zealand showing the six locations sampled for invasive brushtail possums (Trichosurus vulpecula)
Phenotype variation
Brushtail possums in Australia display spatially structured fur colour polymorphism and size variation that has resulted in their treatment as six subspecies, two of which were introduced to New Zealand (Kerle et al. 1991). In southern and eastern Australia (Trichosurus vulpecula vulpecula) the fur of brushtail possums is typically silver-grey above and white cream on the undersides with a black tail and contrasting pale yellow and black marking on the face and ears (Kerle et al. 1991). In Tasmania (T. v. fuliginosus), body size is larger, and the body fur is often almost black, longer and woollier in texture (Kerle et al. 1991; Museums Victoria Sciences Staff 2016). In New Zealand, possum fur traders refer to a range of pelt colours from red-grey to dark brown or black (Nixon 1989). Although copper coloured (red) possums are known from Queensland/Northern Territory of Australia (T. v. johnstonii) there are no records of possums from this region being brought to New Zealand (Hutching 2008).
We used photographs of freshly killed possums to score their fur colour on seven body sections: ear, tail base, shoulders, rump, flank, belly, and flank to belly transition (Fig. 3). For each section we scored predominantly grey characteristics (typical of Trichosurus v. vulpecula) as 1 (e.g. grey shoulder = 1; pale belly = 1), and scored the darkest shades (typical of T. v. fuliginosus) with higher numbers (e.g. black shoulder = 4, black ear = 3). We scored intermediate/mixed colours of brown and red variants (presence of ginger hairs) with intermediate values (e.g. brown ear = 2, red-brown flank = 3; Online Resource 1). The flank to belly colour transition was scored from 1 (sharp) to 3 (continuous). Using the combined scores for all sections each possum was categorised as Grey (score: 1–9), Black (score: 18–23) or a Ginger-mix (score: 10–17) for population genetic analysis (Online Resource 1).

Examples of naturally occurring fur colour variants among feral brushtail possums in Aotearoa. Seven body sections were scored for phenotypic analysis
Population genetic markers
DNA extraction was conducted using the GeneAid™ Tissue DNA Isolation Kit (Genaid Biotech Ltd, Taiwan) following the manufacturer’s instructions with a final elution volume of 200 μl. The quality and quantity of the DNA extraction was assessed using Invitrogen Qubit 4 Fluorometer (ThermoFisher Scientific).
Mitochondrial locus- Polymerase Chain Reaction (PCR) amplification targeted a fragment of the mtDNA D-loop (~ 730 bp) using novel PCR primers designed for the short-eared possum (Trichosurus canninus) and brushtail possum (Trichosurus vulpecula) from whole mtDNA genomes assembled from short read NGS data (Carmelet-Rescan et al. in prep): Tcan_218f (AAGGCAACAACACCTCACCA), Tcan_1049r (AATACGACATCGGCGACCTC), Tvul_228f (CACCTCACCATCAACACCCA) and Tvul_1023r (TCCCGCCCAGTTGATAAACC). Consistent T. vulpecula amplifications and DNA sequences were obtained using the primer combination Tcan_218f and Tvul_1023r. Amplification reactions were in 20 μl volumes with final concentrations of 1 × DreamTaq buffer (ThermoFisher Scientific) and 2.5 mM Magnesium Chloride (additional 0.4 μl of 25 mM MgCl2), 200 μM each deoxy-nucleotide phosphates (dNTP), 0.25 μM each primer, and 0.05U DreamTaq DNA polymerase. Thermocycling used an initial denaturation step at 95 °C for 90 s followed by 36 cycles of 94 °C for 20 s, 51 °C for 20 s, and 72 °C for 1 min; with a final 8-min extension step of 72 °C. Amplification products were sequenced using BigDye® chemistry (Perkin Elmer) following the manufacturer’s protocols on an ABI3730 DNA analyser (Macrogen Inc). Sequences were edited and aligned using the software Geneious 11.5 (Kearse et al. 2012). DNA polymorphism tests, overall haplotype (h) and nucleotide (π) diversity, were estimated using the software DnaSP v 6.12.01. For visualization of these results, a haplotype median-joining network (Bandelt et al. 1999) was inferred using Popart (Leigh and Bryant 2015).
Nuclear loci- Eight microsatellite loci developed for this species were screened using available PCR primers (Taylor and Cooper 1998; Taylor et al. 2004). We used multiplex PCR with four loci in each (M1: Tv_16, Tv_58, Tv_53 and Tv_19 and M2: Tv_64, Tv_PnMs, Tv_M1 and Tv_14) using fluorophores FAM™, VIC™ and TAMRA™ (Online Resource 2). PCR reactions in 10 µl volumes comprised 1 × DreamTaq buffer (ThermoFisher Scientific), 200 µm dNTPs, 0.05U Dream-Taq DNA polymerase, and 0.1 µM each primer. These were subject to initial denaturation at 95 °C for 4 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 45 s and 72 °C for 45 s, and a final extension of 72 °C for 10 min. The amplified microsatellite PCR products were genotyped using a fragment analyser at Macrogen Inc. Allele length was determined using the microsat plugin in Geneious 11.5 (Kearse et al. 2012).
The presence of null alleles and long-allele dropout was examined using the software MicroChecker v2.2.3 (Van Oosterhout et al. 2004). For locus Tv_14 some individuals did not amplify, and the presence of null alleles was suggested by observed homozygotes exceeding the expected (MicroChecker), therefore we removed this locus from further analysis. We tested for linkage disequilibrium using pairwise tests with the software Genepop on the Web (Rousset 2008). Mean number of alleles (Na) at seven loci were estimated in Arlequin 3.5.2.2. Mean gene diversity (G) and mean allelic richness (Ar) were calculated using FSTAT v2.9.4 (Goudet 2003).
Data analyses
Aotearoa New Zealand-wide population structure
We reanalysed an existing dataset obtained for 799 brushtail possums from 14 population samples across Aotearoa (Taylor et al. 2004). If the two subspecies introduced to New Zealand are reproductively partitioned, we expected to see two genotypic clusters within these data. However, if possums had mated randomly with respect to subspecies, we expected to see a mixing of genotypes towards genetic homogeneity. Some spatial differences in genetic composition might remain that reflect initial timing and pattern of genotype introduction. We applied a naïve modelling approach that infers genotypic clusters and assigns individual genotypes using Structure 2.3.2 (Pritchard et al. 2000). Assigned clusters (K) are based on the assumptions that the populations (not the samples) are in Hardy–Weinberg Equilibrium, the loci under analysis are in complete linkage equilibrium, and each cluster identifies as a subset of allele frequencies (Pritchard et al. 2000). The optimal number of population clusters (optimal K) was estimated using the Bayesian Information Criterion.
Bayesian Markov-Chain Monte Carlo (MCMC) clustering of the individual genotypes was run with a the Burnin period set to 5000 and the MCMC repeats thereafter set to 50,000 (Porras-Hurtado et al. 2013). An admixture model was used specifying that the pre-defined populations based on collecting location were not used as priors when assigning individuals to inferred populations. K-values were tested between 1 to 10 for 799 samples with five loci, with 10 iterations for each K. The K-value best fitting the dataset was determined using the Evanno method (Online Resource 3) implemented in Structure Harvester (Earl 2012). To identify cluster modes and visualise population structure inferences across K values, the software Clumpak (Kopelman et al. 2015) was used where the output can be visualised as a stacked bar plot (Fig. 1).
Kenepuru Peninsula population structure
We first looked for evidence that Kenepuru possum samples formed clusters associated with geographical sampling location (five groups for mtDNA locus and six groups for microsatellite loci), and year of sampling (three groups each). Then we looked for an effect of fur colour phenotype on genotypic clustering that would indicate assortative mating. For each division we examined the distribution of haplotypes and genotypes to determine whether there was evidence of a departure from a homogenous population. Normal dispersal behaviour of brushtail possums would be expected to maintain gene flow between locations < 5 kms apart, and therefore geographic structuring of genetic variation within the scale of our Kenepuru sampling would not be expected (Fig. 2). However, a physical impediment to dispersal could limit gene flow across Kenepuru and potentially yield spatially distinct populations, especially if subspecies had been originally introduced to different locations. Temporal genetic structuring could also emerge even over a small number of generations during the 16-year sampling span and would most likely reflect stochastic demographic change if pest control had a significant effect on population size. Positive assortative mating with respect to fur colour phenotypes (subspecies) would maintain ancestral genotypic difference.
To analyse mtDNA haplotype differentiation, 110 individuals were organized into geographical location sets (Table 2) and then grouped according to time (2003, 2016 and 2019). Spatial and temporal population samples were compared using pairwise фST looking for significant deviations from zero (Online Resource 4) assessed using 100 permutations, with a 0.05 level of significance in Arlequin 3.5.2.2 and a Bonferroni correction for multiple tests (0.005 and 0.01667 respectively). Using microsatellite genotypes, tests of Hardy–Weinberg expectations were implemented using GeneAlEx 6.503 (Peakall and Smouse 2006). Deviations from Hardy–Weinberg expected proportions were examined for the full sample of possums from Kenepuru Peninsula (n = 132) and for subsets (including temporal sampling). We used pairwise FST semi-matrix with Slatkin’s method computed in Arlequin 3.5.2.2 (Excoffier and Lischer 2010) to assess population subdivision. Statistically significant departures from zero of pairwise FST values would indicate restricted gene flow (Online Resource 5). The analysis used 100 permutations with a significance level of 0.05 and a Bonferroni correction for multiple tests (0.0033 and 0.01667 respectively).
Fur colour
To explicitly test whether fur colour phenotypes that are associated with distinct subspecies correlate with neutral population genetic structure, 132 Kenepuru possums were classified according to fur colour based on their total score from seven body sections (101 possums were photographed and scored; the remainder were assigned a fur colour at the time of collection without photographs). The mitochondrial diversity of these three phenotype groups was compared using pairwise фST with significance of deviations from zero assessed using 100 permutations, with a 0.05 level of significance in Arlequin 3.5.2.2 and a Bonferroni correction for multiple tests (0.01667). Pairwise FST was estimated for microsatellite genotypes of 132 individuals combined and grouped according to fur colour (as above plus 9 individuals characterised in the field: 66 Grey, 52 Black, 14 Ginger-mix). Statistically significant departures of FST values from zero would indicate non-random mating. We used 100 permutations with a significance level of 0.05 a Bonferroni correction for multiple tests (0.01667). Assortative mating with respect to fur colour would lead to deviations from Hardy–Weinberg proportions so we examined the full dataset and subdivided by fur colour phenotype. We also looked for evidence of genotypic partitioning that would be expected if fur phenotypes were reproductively segregated using the naïve modelling analysis Structure 2.3.2 (Pritchard et al. 2000) with K values of 1 to 3.
Results
Aotearoa New Zealand population structure
Modelling of population genetic structure among 799 brushtail possums from 14 populations across New Zealand with 5 microsatellite loci identified the optimal model had three population clusters (K = 3). Despite the rapid spread of possums across New Zealand, genetic structure within this 2004 sample is readily apparent with most population samples dominated by a single inferred genotype assignment (Fig. 1). However, two population samples included individual possums assigned to all three genotypic clusters present and many uncertain assignments (Waipoua and Shannon, North Island; Fig. 1).
Fur colour variation on Kenepuru Peninsula
We found fur colour traits of the seven body sections to be correlated to varying degrees (Online Resource 6). For example, cream belly and yellow ear always coincided whereas 68% of possums with a grey rump also had pure grey flank (Fig. 3). The frequency distribution of fur colour score in our sample was bimodal, dominated by grey and black individuals with fewer intermediates than expected displaying a mixture of fur colour traits (Fig. 4a). In a sample of 101 Kenepuru possums, we identified 59 pure grey possums with yellow fur behind the ear and grey fur on the tail base, rumps, flanks and shoulders (score: 1–9) and 32 black possums that had black-ears and brown bellies (score: 18–23) (Online Resource 1). Individuals with a mix of grey and black fur were few and typically had ginger hairs on shoulder and flank (score: 10–17) (Fig. 3; Online Resource 1).

Fur colour and genetic variation within a population of brushtail possum (Trichosurus vulpecula) on Kenepuru Peninsula, Aotearoa New Zealand. a Density plot showing the fur colour distribution sampled. b MtDNA haplotype network (684 bp of D-Loop) with fur colour assignment of sampled individuals. Pie size proportional to sample size. c Probability assignment of microsatellite genotypes for the optimal model (K = 2), arranged by fur colour type. d Haplotype network (as above) showing distribution among sample locations, and with codes as given in in Table 2. e Genotype assignment arranged by sampling location
Lack of spatial and temporal genetic structure on Kenepuru Peninsula
D-loop mtDNA sequences of 684 bp were aligned for 110 possum samples and seven haplotypes were identified. The two common haplotypes differed from one another by 2.49% (17 mutations), and five haplotypes observed in one or two possums varied from the common haplotypes by between one and five mutations (maximum divergence 3.2%; Table 2). Overall haplotype diversity (h) was 0.56 and nucleotide diversity (π) was 0.013. Our Koutuwai sample had the highest haplotype diversity (h = 0.71) and nucleotide diversity (π = 0.014). The Manaroa sample (n = 53) had five of the seven haplotypes observed, three of these restricted to the Manaroa sample (Table 2). When grouped geographically into five location samples, none of the 10 pairwise фST estimates from the D-loop locus differed significantly from zero. Similarly, pairwise фST estimates resulting from grouping by year of collection (2003; 2016; 2019) showed no significant deviation from zero (Online Resource 4).
We genotyped 132 possums from Kenepuru using seven microsatellite loci. Total number of alleles per locus ranged from 2 to 13 (Online Resource 2). The Manaroa sample had the highest diversity (Ho = 0.73, He = 0.7) and mean gene diversity (0.71; Table 1). Mean allelic richness per locus ranged between 3.3 (Koutuwai) and 4 (Hopai; Table 1). Significant deviation from Hardy–Weinberg proportions suggested that there was heterozygous excess in the Manaroa sample for just one locus (n = 58; Locus_Tv-58) and heterozygous deficit in the Waitaria Bay population sample for a different locus (n = 12; locus Tv_16; Online Resource 7). Pairwise FST differentiation tests of genotype data showed no significant deviations from zero for any of the pairwise comparisons of location samples. The optimum number of population clusters (K) present in the sample of possums was inferred using Bayesian clustering and the Evanno ∆K comparison approach (Evanno et al. 2005) suggested K = 2. However, K = 2 is effectively the default response from ∆K when there is low signal of structure (Janes et al. 2017) and the distribution of assignment probabilities was consistent with a single genetic population (Online Resource 8; Fig. 4e). When considering sampling time events, only the 2019 sample deviated significantly from Hardy–Weinberg equilibrium at one locus with excess of heterozygous individuals at one locus (n = 56; locus Tv_58; Online Resource 7). Pairwise FST differentiation tests showed no significant deviations from zero when comparing temporal samples (p-value > 0.99 + -0.003).
Genetic structure in relation to fur colour
Based on scoring fur colour of five body sections we classified 132 possums from Kenepuru Peninsula into three colour groups: Grey, Black and Ginger-mix (Table 1). Mitochondrial haplotypes for 110 of these possums found no evidence of genetic differentiation among fur colour groups; pairwise фST did not deviate significantly from zero and there was no concordance of fur and haplotype cluster (Fig. 4b; Online Resource 4). Genotypic data revealed that when samples were combined there were no deviations from Hardy–Weinberg proportions as expected of random mating (chi-squared, p > 0.05). When samples were grouped by fur colour, grey possums had fewer heterozygote individuals than expected at one locus (n = 66; locus Tv_58) and black possums had an excess of heterozygote individuals at one locus (n = 52; locus Tv_M1), while the Ginger-mix possums met Hardy–Weinberg expectations (Online Resource 7). No evidence of genetic differentiation was observed in the nuclear markers with pairwise differentiation; pairwise FST did not deviate significantly from zero. Naive cluster assignment of genotypes showed no support for more than one cluster among brushtail possums on Kenepuru Peninsula. Therefore, our Bayesian clustering analysis did not provide strong evidence of genotypic clustering among brushtail possums from Kenepuru Peninsula (Fig. 4c).
Discussion
Brushtail possums from Australia were released in Aotearoa New Zealand between 1840 and 1900 (Cowan 2005) to establish a fur industry (Pracy 1974). As a result of dispersal from many release sites, the species went on to populate the country, reaching a total population estimated to be approximately 70 million at the end of 1900s (Clout 2000). Brushtail possum invasions led to the drastic decline of native flora and fauna (Sadlier 2000), and possums emerged as prominent reservoirs of bovine tuberculosis in New Zealand (Livingstone et al. 2015). Control measures targeting feral possums are spatially and temporally uneven, potentially contributing to a complex and poorly understood landscape genetic structure. As genetic composition of invasive populations can have a strong influence on their invasion characteristics and responses to control measures (Lee 2002), developing a model of gene flow and identifying eradication units within Aotearoa possums is essential for their future control and eradication (Adams et al. 2014; Tompkins 2018).
Feral brushtail possums are genetically diverse at neutral loci (Triggs and Green 1989; Taylor et al. 2004). Reanalysis of microsatellite data from nation-wide sampling (Taylor et al. 2004) revealed partitioning of diversity within the species into at least three genetic clusters. However, the complex history of possum introductions and pattern of subsequent translocation and spread cannot be fully elucidated with this scale of sampling and five nuclear loci. Additional markers will provide resolution for a national wide study of this species’ invasion.
Notably the brushtail possums in Aotearoa New Zealand exhibit fur colour variation that distinguishes the two subspecies of Trichosurus vulpecula introduced to the country (Fig. 1). This raises the possibility that regionally partitioned lineages might have distinct traits and that invasive populations of mixed ancestry may have novel genotypes (e.g. Lu et al. 2011; Smith et al. 2020). However, the fine-scale patterns of gene flow in this invaded landscape remain largely concealed. Analysis of neutral genetic loci and phenotypic information found evidence that distinct brushtail possum lineages established in the North Island of New Zealand from separate introductions from Australia have remained partitioned (Sarre et al. 2014; Campbell et al. 2021). These analyses imply ongoing assortative mating of genotypes characterised by different fur colour (and origin), highlighting the potential for cryptic lineages (Bastos et al. 2011; Jarić et al. 2019; Perry et al. 2018).
We conducted a fine-scale analysis of genetic structure of brushtail possums collected from Kenepuru Peninsula (Fig. 2), which is a near closed system with a barrier to dispersal from the mainland in the form of a narrow isthmus. Within this landscape we sought evidence of genetic partitioning using seven nuclear microsatellite markers and a mitochondrial marker to resolve reproductive exchange among possums of mixed ancestry. Phenotypic variation based on morphological data suggested a preponderance of grey and black types with a deficiency of intermediates. However, our genetic analyses using mitochondrial markers showed no association of haplotype with fur colour, and pairwise differentiation tests using nuclear loci showed no correlation with fur colour (Fig. 3). Our results contrast with previous evidence for assortative mating (Sarre et al. 2014) and the presence of a contact zone linked to the genotypic differences between two populations of possums. The stepped cline detected in the eastern North Island of New Zealand could be due to spatial effects such as the presence of a large river rather than a mating bias. Priority effects established by separate historic introductions of two different lineages of possum in separate parts of Hawke’s Bay may be sufficient to explain the data. Numerical dominance on either side of a river could constrain gene flow between lineages. In contrast, Kenepuru Peninsula lacks major landscape barriers allowing complete mixing of source populations that must have included alleles from both subspecies, thus showing no evidence to support different fur colours belonging to different genotypic clusters.
We documented for the first time mtDNA haplotype variation among the brushtail possum in New Zealand (Table 2) and identified a total of seven haplotypes (Fig. 4d), with a maximum of 3.2% sequence divergence. Introduction from different source populations in Australia most probably explains the presence of two distinct mtDNA lineages within the Kenepuru Peninsula population (Carmelet-Rescan et al. in prep). Despite rich diversity at nuclear loci (Table 1), our samples were all from the same population, as revealed by the estimates of pairwise differentiation for both sets of markers (фST and FST = 0). Possums on the Kenepuru Peninsula are not spatially structured and there seems to be no spatial restrictions to gene flow over this area. This agrees with other estimates of eradication units spanning about 20 kms (Adams et al. 2014) and estimates that 20–30% of juvenile possums will disperse up to five kms (Cowan and Clout 2000). Dispersal routes are important in terms of local management, as local eradication is likely to increase opportunities for possum reinvasion, possible in the absence of external obstacles. Increased dispersal and gene flow could generate novel genotypes, enhance evolutionary potential, spread pest control resistance traits and increase disease transmission (Allendorf and Leary 1986; Byers et al. 2015).
We conclude that possums on the Kenepuru Peninsula function as a single population despite a deficit of individuals with an intermediate fur colour. Our genetic data provide enough evidence to refute the hypothesis of assortative mating segregating possum subspecies, which substantially simplifies their management. Predator Free 2050 aspires to rid Aotearoa New Zealand of possums and mustelids by the year 2050 (Tompkins 2018). To do this we need to consider factors that might reduce the effectiveness of management strategies including identification of eradication units, reinvasion pathways and the potential of hybrid advantage.
Data availability
All authors are aware of the use of materials and software used support the claims of this study. mtDNA sequences and microsatellite genotypes are available during the review stage from: http://evolves.massey.ac.nz/Toolkit.htm. When accepted for publication all data will be uploaded to Dryad.
References
Adams AL, Van Heezik Y, Dickinson KJM, Robertson BC (2014) Identifying eradication units in an invasive mammalian pest species. Biol Invasions 16:1481–1496
Allendorf FW, Leary RF (1986) Heterozygosity and fitness in natural populations of animals. Conserv Biol Sci Scarcity Divers 57:58–72
Atkinson I, Campbell D, Fitzgerald B et al. (1995) Possums and possum control; effects on lowland forest ecosystems. A literature review with specific reference to the use of 1080. p 32
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bastos AD, Nair D, Taylor PJ et al (2011) Genetic monitoring detects an overlooked cryptic species and reveals the diversity and distribution of three invasive Rattus congeners in South Africa. BMC Genet 12:26
Benvenuto C, Cheyppe-Buchmann S et al (2012) Intraspecific hybridization, life history strategies and potential invasion success in a parasitoid wasp. Evol Ecol 26:1311–1329
Byers JE, Smith RS, Pringle JM et al (2015) Invasion expansion: time since introduction best predicts global ranges of marine invaders. Sci Rep 5:12436
Caley P, Hickling G, Cowan P et al (1999) Effects of sustained control of brushtail possums on levels of Mycobacterium bovis infection in cattle and brushtail possum populations from Hohotaka, New Zealand. N Z Vet J 47:133–142
Campbell CD, Cowan P, Gruber B et al. (2021) Has the introduction of two subspecies generated dispersal barriers among invasive possums in New Zealand?. Biol Invasions, pp 1–15
Clout M (2000) Anatomy of a disastrous success: the brushtail possum as an invasive species. In: Montague TL (ed) The brushtail possum-biology, impact and management of an introduced marsupial. Manaki Whenua Press, Lincoln, pp 1–9
Cowan P (2005) Brushtail possum. In: King CM (ed) The handbook of New Zealand mammals. Oxford University Press, Melbourne, pp 56–80
Cowan P, Clout M (2000) Possums on the move: activity patterns, home ranges, and dispersal. In: Montague TL (ed) The brushtail possum: biology, impact and management of an introduced marsupial. Manaaki Whenua Press, Lincoln, pp 24–34
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eason C, Miller A, Ogilvie S et al (2011) An updated review of the toxicology and ecotoxicology of sodium fluoroacetate (1080) in relation to its use as a pest control tool in New Zealand. N Z J Ecol 35:1–20
Efford M (2000) Possum density, population structure, and dynamics. In: Montague TL (ed) The brushtail possum-biology, impact and management of an introduced marsupial. Manaki Whenua Press, Lincoln, pp 47–61
Engler JO, Sacher T, Coppack T et al (2019) Assortative mating frames establishment in a young island bird population. R Soc Open Sci 6:190050
Estoup A, Ravigné V, Hufbauer R et al (2016) Is there a genetic paradox of biological invasion? Annu Rev Ecol Evol Syst 47:51–72
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Goldson S, Bourdôt G, Brockerhoff E et al (2015) New Zealand pest management: current and future challenges. J R Soc N Z 45:31–58
Goudet J (2003) FSTAT (Version 2.9. 4), a program (for windows 95 and above) to estimate and test population genetics parameters. Department of Ecology & Evolution, Lausanne University, Switzerland, p 53
How R, Kerle J (1995) Common brushtail possum. In: Strahan R (ed) The Mammals of Australia, pp 273–275
Hutching G (2008) Possums. In: Te Ara - the Encyclopedia of New Zealand. http://www.TeAra.govt.nz/en/possums/print
Janes JK, Miller JM, Dupuis JR et al (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Jarić I, Heger T, Monzon F et al (2019) Crypticity in biological invasions. Trends Ecol Evol 34:291–302
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kerle J, McKay G, Sharman G (1991) A systematic analysis of the brushtail possum, Trichosurus-vulpecula (Kerr, 1792) (Marsupialia, Phalangeridae). Aust J Zool 39:313–331
Kopelman NM, Mayzel J, Jakobsson M et al (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:368–391
Leigh JW, Bryant D (2015) popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Livingstone P, Hancox N, Nugent G et al (2015) Toward eradication: the effect of Mycobacterium bovis infection in wildlife on the evolution and future direction of bovine tuberculosis management in New Zealand. N Z Vet J 63:4–18
Lu M, Wingfield MJ, Gillette N et al (2011) Do novel genotypes drive the success of an invasive bark beetle–fungus complex? Implications for potential reinvasion. Ecology 92:2013–2019
Museums Victoria Sciences Staff (2016) Trichosurus vulpecula Common Brushtail Possum in Museums Victoria Collections. https://collections.museumsvictoria.com.au/species/8449. Accessed 16 July 2020
Nixon A (1989) Fur growth and replacement in the brushtail possum, Trichosurus vulpecula Kerr. Unpublished PhD thesis, Massey University, Palmerston North
Parkes J, Murphy E (2003) Management of introduced mammals in New Zealand. NZ J Zool 30:335–359
Paulay G (1994) Biodiversity on oceanic islands: its origin and extinction1. Am Zool 34:134–144
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Perry KD, Baker GJ, Powis KJ et al (2018) Cryptic Plutella species show deep divergence despite the capacity to hybridize. BMC Evol Biol 18:77
Porras-Hurtado L, Ruiz Y, Santos C et al (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 4:98
Pracy LT (1974) Introduction and liberation of the opossum (Trichosurus vulpecula) into New Zealand. New Zealand Forest Service, Wellington
Prior KM, Adams DC, Klepzig KD et al (2018) When does invasive species removal lead to ecological recovery? Implications for management success. Biol Invasions 20:267–283
Pritchard JK, Stephens M, Rosenberg NA et al (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Quilodrán CS, Austerlitz F, Currat M et al (2018) Cryptic biological invasions: a general model of hybridization. Sci Rep 8:1–12
Ranke PS, Skjelseth S, Hagen IJ et al (2020) Multi-generational genetic consequences of reinforcement in a bird metapopulation. Conserv Genet 21:1–10
Rius M, Darling JA (2014) How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol Evol 29:233–242
Rouco C, Norbury GL, Smith J et al (2013) Population density estimates of brushtail possums (Trichosurus vulpecula) in dry grassland in New Zealand. N Z J Ecol 37:12–17
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Sadleir RMFS (2000) Evidence of possums as predators of native animals. In: Montague TL (ed) The brushtail possum: biology, impact and management of an introduced marsupial. Manaaki Whenua Press, Lincoln, pp 126–131
Sarre SD, Aitken N, Adamack AT et al (2014) Creating new evolutionary pathways through bioinvasion: the population genetics of brushtail possums in New Zealand. Mol Ecol 23:3419–3433
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Smith AL, Hodkinson TR, Villellas J et al (2020) Global gene flow releases invasive plants from environmental constraints on genetic diversity. Proc Natl Acad Sci 117:4218–4227
Spatz DR, Zilliacus KM, Holmes ND et al (2017) Globally threatened vertebrates on islands with invasive species. Sci Adv 3:e1603080
Sullivan LL, Li B, Miller TEX, Neubert MG, Shaw AK (2017) Density dependence and fluctuating invasion speeds. PNAS 114:5053–5058
Taylor A, Cooper D (1998) Microsatellite markers for the Phalangerid marsupial, the common brushtail possum (Trichosurus vulpecula). Mol Ecol 7:1780–1782
Taylor A, Cowan P, Fricke B et al (2004) High microsatellite diversity and differential structuring among populations of the introduced common brushtail possum, Trichosurus vulpecula, in New Zealand. Genet Res 83:101–111
Thomson G (1922) The naturalisation of plants and animals in New Zealand. Cambridge University
Tompkins DM (2018) The Research Strategy for a ‘Predator Free’ New Zealand. In Proc Vertebr Pest Conf 28:11–18
Trewick S, Bland K (2012) Fire and slice: palaeogeography for biogeography at New Zealand’s North Island/South Island juncture. J R Soc N Z 42:153–183
Triggs S, Green W (1989) Geographic patterns of genetic variation in brushtail possums Trichosurus vulpecula and implications for pest control. N Z J Ecol 12:1–10
Van Oosterhout C, Hutchinson WF, Wills DP et al (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wodzicki K, Wright S (1984) Introduced birds and mammals in New Zealand and their effect on the environment. Tuatara (wellington) 27:77–104
Woinarski JC, Burbidge AA, Harrison PL (2015) Ongoing unraveling of a continental fauna: decline and extinction of Australian mammals since European settlement. Proc Natl Acad Sci 112:4531–4540
Acknowledgements
This work was supported by funding from Operational Solutions for Primary Industries New Zealand Ltd (OSPRI). Sampling of brushtail possums on Kenepuru Peninsula was assisted by Steve Podjursky (Bowron, Jackson and Harvey properties 2016), Emily Osbourne and Mitch Bartlett (Skiddaw property, Koutuwai 2019), Alistair Jenner (Millig and Waitaria Bay properties 2019), Kristen and Mike Gerard (Hopai and Port Gore properties 2019), and Landcare Research (Kenepuru, 2003). We thank Andrea Taylor and team for making raw data from the 2004 possum study available for reanalysis.
Funding
This research was funded by Operational Solutions for Primary Industries New Zealand Ltd (OSPRI), Predator Free 2050 Limited and Massey University Research Fund (to SAT).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Tissue samples and photographs were obtained by Ralph Powlesland. Data generation and analysis was conducted by Nimeshika Pattabiraman with the assistance of Mary Morgan-Richards and Steve Trewick. Writing was led by Nimeshika Pattabiraman and supported by all authors. Figures were developed by Steve Trewick. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pattabiraman, N., Morgan-Richards, M., Powlesland, R. et al. Unrestricted gene flow between two subspecies of translocated brushtail possums (Trichosurus vulpecula) in Aotearoa New Zealand. Biol Invasions 24, 247–260 (2022). https://doi.org/10.1007/s10530-021-02635-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-021-02635-z