
Abstract
Purpose
The efficacy of drugs against Salmonella infection have compromised due to emerging XDR H58 strain. There is a dire need to find novel antimicrobial drug targets as well as drug candidates to cure by the XDR strain of Salmonella. It is observed that the complete genome sequence of the XDR H58 strain contains a large number of hypothetical proteins with unknown cellular and biological functions. Hence, it is indispensable to annotate these proteins functionally as well as structurally to identify novel drug targets.
Methods
In the current study, a comparative genomics and proteomics based approach was applied to find the novel drug targets in XDR strain while comparing the MDR and NR strains of Salmonella typhi.
Results
The characterization of ~ 350 hypothetical proteins were performed through determination of their physio-chemical properties, sub-cellular localization, functional annotation, and structure-based studies. As a result, only five proteins were prioritized as essential, druggable, and virulent proteins. Moreover, only one protein i.e. WP_000916613.1 was functionally annotated with high confidence and subjected to further structure-based analysis.
Conclusion
The current study presents a hypothetical protein from the XDR S. typhi proteome as a potential pharmacological target against which novel therapeutic candidates may be predicted. The outcome of the current study may lead to formulate a general set of pipelines for better understanding of the role of hypothetical proteins in pathogenesis of not only Salmonella but also for other pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salmonella enterica subsp. serovar typhi (S. typhi) is a rod-shaped, Gram-negative, and motile bacterium. It is characterized as human-restricted monophyletic serovar that is found in humans only (Feasey et al. 2015; Thanh et al. 2016). It is responsible for a large health burden globally i.e. typhoid fever, which is one of the life-threatening illness. Typhoid fever remains a significant illness that attributes to an estimate of 21.6–26.9 million cases and 216,000 deaths each year (Klemm et al. 2018). It is the most common illness that occurs in Pakistan i.e. over 100,000 cases reported every year (Sah et al. 2020). Alarmingly, in November 2016, Pakistan health authorities have reported an ongoing outbreak of newly emerged Extensively Drug Resistant (XDR) strain S. typhi, haplotype 58 (H58) that was spread in the Hyderabad district of Sindh province (Akram et al. 2020). The Year of Life Lost’s (YLLs) has estimated that 10.9 million cases of typhoid fever and 116.8 thousand deaths were caused by the newly emerged H58 XDR Salmonella typhi (Ali et al. 2016). Frighteningly, the increasing incidence of H58 and its dominance over other S. typhi strains have been reported worldwide in Australia, Canada, Denmark, Taiwan, Ireland, the United Kingdom and the United States (Akram et al. 2020). Unfortunately, there is no official data on the prevalence of XDR S. typhi outside of Sindh province, though there are cases reported from all around the country. During the current SARS-CoV-2 pandemic, a substantial number of typhoid cases have emerged with clinical signs that are identical to COVID-19. Furthermore, > 20,000 typhoid cases were recorded in Pakistan only in June 2020 (Rasheed et al. 2020).
Unfortunately, the current treatment for typhoid fever (i.e. chloramphenicol, ampicillin, and trimethoprim-sulfamethoxazole—first-generation antibiotics) is endangered due to the emergence of S. typhi haplotype 58. The S. typhi halplotype 58 is associated to MDR resistance to second-generation (i.e. ampicillin, chloramphenicol, trimethoprim-sulfamethoxazole ciprofloxacin, streptomycin, tetracycline and fluoroquinolones) as well as third-generation antibiotics (i.e. cephalosporins) (Ali et al. 2016; Klemm et al. 2018). The rise in the resistance of H58 clonal strain was observed in numerous parts of Pakistan increasing the potential risk at all three levels (i.e. Global, National, and Regional) (Ali et al. 2016). As therapeutic options are severely limited, the potential for further spread is a serious concern (Rasheed et al. 2020).
The Whole Genome Sequencing (WGS) has traditionally been used to track bacterial and viral disease distribution and also to investigate the evolution of antibiotic resistance mechanisms. Isolated cases of XDR S. typhi from Taiwan, Canada, and Denmark have shown a close connection (100 percent nucleotide match) to the closest sequenced XDR strain from Pakistan. This emphasizes the current outbreak's global impact. The fact that any SNPs discovered across S. typhi isolates during an outbreak suggest prolonged transmission (Rasheed et al. 2020). Although whole-genome sequencing helped to understand the blueprint of the life and identification of numerous open reading frames with enough evidence for gene expression (Jamilah et al. 2020; Wadood et al. 2018). However, it's still difficult to assign the function to all the genes due to the lack of of protein sequences with annotated biochemical function (Sivashankari and Shanmughavel 2006). This process is itself time-consuming, costly and despite several efforts, only 50% of genes are annotated, leaving considerable amount of protein functionally unpredictable and are classified as conserved hypothetical proteins (homogeneous to unknown gene function) or hypothetical protein (no known homologous) (Sivashankari and Shanmughavel 2006). Thus, the determination of protein function is still the challenge in post genomic era. This demands the bioinformatics methods to predict functions of un-annotated protein sequence by developing efficient tools (da Costa et al. 2018). During genome analysis, these hypothetical proteins are predicted by various software as a large open reading frame without a characterized homologue in the protein database. It returns "hypothetical protein" as an annotation remark (da Costa et al. 2018; Pranavathiyani et al. 2020). It is vital to understand the function of hypothetical proteins as many of them might be associated with the disease conditions. Upon investigation, it is predicted that these hypothetical proteins also tend to serve as drug targets because of their important role in the biochemical, physiological pathways and may act as biomarkers, pharmacological targets in proteomic and genomic research. Several methods are being used to identify and assign the function to hypothetical proteins such as the domain homology search, homology modeling and structure prediction with biochemical function assessment. Certainly, among these approaches “Subtractive Genomics Approach” is one of the most widely applied methodology to prioritize the drug targets. In silico strategies are cost effective and fast enough to annotate hypothetical proteins and explore their functions. The in silico approaches to the functional prediction of hypothetical proteins have been successfully used for several bacterial species such as vibrio cholera (Islam et al. 2015), Neisseria gonorrhea (Bhairamadgi and Katti 2013), Clostridium difficile (Dannheim et al. 2017), Candida dubliniensis and Staphylococcus aureus (Varma et al. 2015). Therefore, the purpose of this work is to assign the function to the hypothetical proteins present in the genome of the XDR strain of S. typhi (H58) with a comparative analysis to MDR (343077_213147 isolated from Bangladesh, and CT-18) and sensitive strains (Ty2 and STyphi_1553) specific to South Asia and globally Known. The main goal of this study was to better comprehend the hypothetical proteins S. typhi by assigning structural and biological functions to them. Comparative proteomic and genomic analysis, subtractive proteomic and genomic approach, functional annotations, and physico-chemical properties analysis was performed, and subcellular distribution, secondary structure, and active site were predicted. Furthermore, using structure based techniques, a reasonable quality model of the shortlisted protein was generated. The comparative analysis of these five strains led to novel proteins that contribute to the adaptation of bacterium to such harsh conditions. Several bioinformatics tools are used in this work for the functional annotation of such proteins, which might lead to the discovery of new therapeutic targets for screening, drug development, and design for the treatment against the Salmonella infection.
Material and methods
In the current study, the structural and functional annotation of hypothetical proteins of the XDR H58 Salmonella typhi was carried out. The work flow was performed by employing a comparative subtractive proteo-genomics in four phases, i.e. Comparative Proteo-genomic Analysis, Functional Annotation, Subtractive Genomic Phase, and Structure based studies as shown in Fig. 1. Various bioinformatics methods along with different algorithms were used for the annotation and functional characterization as shown in Fig. 2.

Complete flowchart of proposed study for functional and structural annotations of hypothetical proteins inferring potential drug targets

Flowchart showing all tools used for functional and structural annotation in the current study
Phase 1: comparative proteo-genomic analysis
This approach is based on the comparative analysis of the proteome data of hypothetical proteins from five strains of Salmonella typhi. The objective was to find the proteins uniquely present in XDR strain. For that purpose, the comparative analysis of XDR H58 strain with two MDR strains and two sensitive strains (specific to Asia and globally Known) was performed.
Data collection of genome and proteome
The XDR strain H58, MDR strains specific to Asia and globally known, and non-resistant strains specific to Asia and globally prevalent strains (both Genome and Proteome) were retrieved from the National Center for Biotechnology Information (i.e. RefSeq NCBI) (Table 1) (Pruitt et al. 2005). Whereas human proteome was retrieved from Universal Protein Resource (UniProt) database (Consortium 2015). The Database of Essential Genes (DEG) (Zhang et al. 2004) was used to investigate the essentiality of the drug targets. Furthermore, the Drug Bank database was used to assess the druggability of the shortlisted targets (Wishart et al. 2018).
Retrieval of Homo sapiens proteome
The complete proteome of Homo sapiens (human) was retrieved from the Universal Protein Resource database (https://www.uniprot.org/uniprot/query=homo+sapiens). The proteome size of Homo sapiens was ~ 20,318 proteins (Uddin and Saeed 2014) with an accession ID: UP000005640.
Retrieval of salmonella typhi hypothetical proteins
Complete proteomes of XDR, MDR, and NR strains were obtained from the NCBI FTP to fetch hypothetical proteins. The proteome size of XDR, MDR, and NR strains was consisted of ~ 4500 proteins in each of the five strains. It was observed that XDR strain contains ~ 535 hypothetical proteins that corresponds to the ~ 11.8% of its whole proteome size (Fig. 3). These hypothetical proteins were mined and retrieved through their NCBI IDs. The detail of strains and proteins is illustrated in Table 1.

Complete proteome of XDR H58 Salmonella strain showing hypothetical proteins
Comparative genomic analysis of XDR, MDR, and NR strains
Eventually, the hypothetical proteins from MDR and NR strains were compared with the XDR strain to identify the novel proteins and genes that are only present in XDR strain. The CgView tool was used for the genome alignment of these five strains of Salmonella typhi to find unique as well as novel XDR H58 proteins (Grant and Stothard 2008).
Phase 2: structural and functional annotation of identified hypothetical proteins
Annotation of functional domain and protein superfamily
The proteins were submitted for annotation using Argot2 (Falda et al. 2012), PFP (Hawkins et al. 2009) and ProtoNet (Sasson et al. 2003) using a threshold E-value of 10–5 to classify shortlisted Hypothetical Proteins (HPs) into functional families based on their sequences, structures and functions (Fig. 2). Each tool predicts function of protein based on specific algorithms and provides scores of confidentialities in predicted results e.g., Very high confidence is > 20 k, High confidence is > 10 k, Moderate confidence is > 500, Low confidence is ≥ 100, and below low confidence is < 100 for PFP tool. Whereas threshold was set as ≥ 200 for Argot2 tool. According to the Argot2, PFP, and ProtoNet scores, any HP-presenting products that described family and/or protein domains were chosen for further study based on available bioinformatics tools for domain and function assignment i.e., SMART (Schultz et al. 1998) using a default parameters.
Performance assessment
The predicted function of shortlisted proteins of XDR strains through functional annotation tool was validated using Receiver Operating Characteristics (ROC) analysis. For that purpose, a Web-based calculator easyROC was used, which is a web-tool for ROC curve analysis (Goksuluk et al. 2016). The function of 100 proteins (already annotated through sequencing) was predicted from Salmonella typhi using ROC analysis in order to validate the efficiency of the tool (da Costa et al. 2018). The integers “2”, “3”, “4”, and “5” were given as confidence rating based on scores generated through each tool e.g. 2 is Probably negative, 3 is Possibly negative, 4 is Possibly positive, and 5 is Probably positive. The binary numerals “0” or “1” were given to classify the predictions as true positive (Definitely positive) (“1”) or true negative (Definitely negative) (“0”) (da Costa et al. 2018).
Physico-chemical properties
In a biological system, all proteins are dependent on their structures and activities, which are in turn dependent on physical and chemical parameters. The physico-chemical properties of shortlisted hypothetical proteins were predicted through ProtParam tool (Garg et al. 2016). The output of the ProtParam indicates multiple variables associated to the physical and chemical properties related to proteins. The physicochemical properties are consisting of molecular weight, pK values of different amino acids instability index, GRAVY values, isoelectric pH, hydropathicity, approximate half-life of HPs and aliphatic index of the HPs.
Phase 3: comparative subtractive proteo-genomics approach
The Comparative Subtractive Genomics is a powerful approach that applies the comparative analysis of different strains and the sequence subtraction between the host and the pathogen (Fenoll et al. 2009). Thus, providing necessary information for a set of genes/proteins essential to the microorganism but not existing in the respective host (Supplementary Data S1/Figure S1) (Uddin et al. 2020). The subtractive genomics plays a role of great importance in potential drug target identification as unique and essential to the pathogen survival without altering the host (human) systemic mechanism and pathways (Uddin and Saeed 2014).
Non-homologous protein identification
Accordingly, the HPs only found in XDR strain were subjected to BLASTp with a cut-off value of E-value 10−4 against whole proteome of Homo sapiens to identify the unique proteins that are non-homologous to the human. The BLASTp resulted in ‘Hits’ (Homologous sequence between the host and the pathogen) and ‘No Hits’ (Non-homologous sequences). The ‘No Hits’ proteins were selected for further steps of the study (Uddin and Azam 2019). The non-homologous (No Hits) sequences were selected and retrieved for further analysis to avoid the functional and structural similarities with human proteins in order to minimize the cross reactivity.
Essential non-homologous genes identification
The proteins that play a major role in cellular metabolisms are said to be essential for any organism’s survival (Deng et al. 2011). Thus, a BLASTp of non-homolog hypothetical proteins was performed against DEG with cut-off E-value 10–5 to shortlist proteins essential to the pathogen’s survival. The significant similar sequences were retrieved for further analysis and the remaining non-similar proteins were excluded.
Druggability of essential protein
Furthermore, the essential non-homolog proteins were assessed through BLASTp with E-value 10−3 against the Drug Bank database to determine their drug target like ability and finally classified them as novel drug targets. The ‘Hits’ are proteins with high similarity frequency (80% or more) to the FDA approved DrugBank database were considered as druggable target and therefore, selected for further analysis.
Hypothetical protein’s virulence prediction
The understanding of pathogenic-ability of micro-organisms can be estimated through identification of virulent proteins from its sequenced genome (Gupta et al. 2014). The identification of such proteins can provide valuable information of virulence by comparing the metagenome of healthy and diseased individuals and estimating the proportion of pathogenic species. The virulence of hypothetical proteins were predicted through the MP3 database (Gupta et al. 2014). All protein sequences were submitted to the MP3 database that classified the virulent and non-virulent proteins.
Sub-cellular location prediction
The subcellular location of final shortlisted protein targets was determined by using PSORTb v.3.0. and CELLO2GO tool (C.-S. Yu et al. 2014) to get information about HPs localization. These tools predict the sub-cellular localization i.e. presence of proteins in cell organelles which determines whether a protein is present in the cell membrane, cytoplasm, extracellular, inner membrane, outermembrane, periplasmic proteins, and unknown regions.
Phase 4: structure based studies of identified hypothetical proteins
Protein structure prediction and validation
The structure-based drug design requires the understanding of protein’s molecular function and its 3D structure. The proteins shortlisted from the above steps were searched for their structures in PDB. A suitable template for the protein structure modeling was found through BLASTp. Furthermore, the 3D structures of shortlisted drug targets were modeled by homology using the Modeler server (Fiser and Šali 2003).
The validation of modeled structure is required for further structure-based studies i.e., molecular docking. The modeled structure was validated through PROCHECK using a cut-off value as > 90% of residues in favorable regions of Ramachandran plot and PSIPRED (McGuffin et al. 2000). Both tools verified the modeled structure of the proteins based on their respective principles (i.e. stereochemical quality and secondary sequences alignment, respectively). The structure analysis has more values in predicting the function of a protein than sequence-based methods. It is because the homologous proteins show more conservation in function during the evolution process. The ProFunc tool was used for functional validation through structure (Laskowski et al. 2005).
Metabolic pathway analysis of shortlisted proteins
In this step, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used for metabolic pathways retrieval of shortlisted hypothetical proteins from XDR Salmonella typhi strain using the KAAS server. The protein sequences belonged to the unique metabolic pathways of S. typhi were retrieved from the NCBI database for further analysis.
Phylogenetic analysis
Moreover, the evolutionary relationship and interrelationship of shortlisted hypothetical proteins between other organisms was known by generating phylogenetic tree. The phylogeny of hypothetical proteins was generated through Clustal Omega (Sievers et al. 2011) and was visualized through the iTOL tool (Letunic and Bork 2007).
Active site prediction
Finding the active site of a protein where a ligand could bind to alter its function is a crucial step during the protein’s structure modeling. Therefore, the DoGSite Scorer tool was used for this purpose of locating the binding/active site of the protein using a cut-off value of 1.0 (Volkamer et al. 2012). The DoGSite Scorer identifies active site pockets based on the physio-chemical properties of the protein residues. The predicted active site can be used to dock ligand against respective proteins.
Ligand prediction
The ligand for the shortlisted protein was not reported earlier in the literature. Therefore, ProBis server was used to find the potential ligands. The ProBis examines the fundamental interactions between ligand and the drug targets. It identifies suitable ligands for proteins by searching a best annotated protein structure in PDB database along with its ligand for the template protein (Konc and Janežič 2010). On the basis of the proposed ligand, one may design new drugs in further drug discovery stages. The ProBis server is freely available at http://probis.cmm.ki.si.
Molecular docking studies
In molecular docking the most effective ligand shows the minimal sore of docking for its target proteins. The proteins with modeled structure were used as target protein while identified compound was used as a ligand. The standard docking procedure was used for the molecular docking using AutoDock (Uddin and Azam 2019).
Post docking analysis
Furthermore, the interaction of docked protein–ligand complexes were analyzed through the PoseView program (Stierand and Rarey 2010). The hydrogen bonding and hydrophobic interactions between the receptor and ligand atoms were identified within a range of 5 Å and visualized through Chimera tool, respectively (Pettersen et al. 2004).
Results
Phase 1: comparative proteo-genomic analysis
Genome and proteome comparison between XDR, MDR, and NR strains of S. typhi
The identified 535 HPs from the S. typhi were retrieved through their NCBI IDs and later compared against the whole genome of MDRs and NR (global and Asian) strains through CgView tool. It aligned these five strains (XDR strain HPs, MDRs and NRs) based on BLAST and Multiple Sequence Alignment (MSA) algorithm to identified the novelty of HPs uniquely found in XDR strain. The results showed ~ 350 notable number of novel hypothetical proteins observed uniquely in XDR as shown in Fig. 4. In order to further validate the predicted CgView, the whole proteome of five strains (XDR, MDRs, and NRs) was subjected to BLASTp, and 'No Hits’ were retrieved. Similarly, among 535 hypothetical proteins of XDR Salmonella typhi, 351 hypothetical proteins were uniquely present in the XDR strain i.e. absent in other four strains. These 351 hypothetical proteins were selected for further downstream analysis.

The comparative analysis of all five strains showing hypothetical genome alignments
Phase 2: functional characterization of hypothetical proteins
Functional family classification of shortlisted proteins
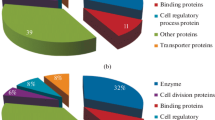
The existence of a certain amino acid and its occurrence in the main sequence, which is also a guiding factor in the three-dimensional (3D) structure of a protein, determines its function. The sequence-based search for shortlisted 351 proteins was performed using PFP (Hawkins et al. 2009), Argot2 (Falda et al. 2012), ProtoNet (Sasson et al. 2003), and SMART tools (Schultz et al. 1998) to obtain extensive information about comparable 3D structures and other associated factors such as class and family. The PFP tool predicted the function in terms of molecular, biological, and cellular component with the confidence score in predicted functions. It mainly predicted the HPs as NADP binding, ATP binding, metal ion binding protein and as ligases. The Argot2 tool predicted function of hypothetical proteins in terms of molecular, biological, and cellular components. Among the 351 hypothetical proteins, it predicted functions for ninety-eight hypothetical proteins and characterized as Endonuclease, Dehydrogenase, and structural proteins. The function of shortlisted hypothetical proteins was also identified by the ProtoNet tool to confirm the predicted function by other tools. It predicts the hypothetical protein function based on cluster network identification in terms of molecular function and by InterPro family. The ProtNet classified hypothetical proteins as Endonucleases, Oxideoxyribonuclease and as Ribosomal proteins. Whereas, the SMART tool was used for the prediction of functional domains of hypothetical proteins. It predicted only transmembrane functional domains for ~ 60 hypothetical proteins. The predicted function for the hypothetical proteins through these tools is enlisted in Supplementary Data S2.
Performance assessment of tools used for functional annotation
As described above, the confidence integers used for ROC analysis were “2”, “3”, “4”, and “5” based on their scores while true positives and true negatives were denoted by binary number i.e. “1” and “0”, respectively (Supplementary Data S3). These data of classification were subjected to an online ROC analysis called easyROC tool, which estimated the sensitivity, accuracy, specificity, and the ROC area of the functional predictions of hypothetical proteins (as shown in Table 2). The average accuracy obtained by the applied pipeline was 91.3% (Fig. 5). The results from the ROC analysis indicated the high reliability of the set of bioinformatics tools used in the present study.

ROC plot presenting the change of trend of specificity, sensitivity at 100 proteins size sample for PFP, ArGot2, and ProtoNet, respectively
Physicochemical properties prediction
The computed physicochemical properties of all 351 shortlisted hypothetical proteins indicated the molecular weight, isoelectric point, extinction coefficient, instability index, aliphatic index, and Grand Average of Hydropathicity (GRAVY) for each protein (Garg et al. 2016). The Supplementary Data S4 showed the predicted results for all hypothetical proteins. It was observed through the ProtParam analysis that only 142 proteins were found to be stable.
Phase 3: subtractive proteo-genomic analysis
Non-homologous hypothetical proteins identification
As described above, total of 351 hypothetical proteins were retrieved from the XDR strains. These 351 proteins were subjected to BLASTp to determine non-homologue protein sequences against the human host proteome. The result revealed 350 proteins as non-homologous proteins i.e. present only in the XDR strain of Salmonella typhi. These proteins were further analyzed in subsequent steps.
Druggability of therapeutic targets
Eventually, the shortlisted 350 non-homologous proteins were subjected to BLASTp to analyze the drug target like ability against the DrugBank database. The BLASTp search resulted in eight XDR hypothetical proteins as the druggable proteins.
Identification of essential proteins
The essentiality of all eight drug-target like proteins were determined by performing BLASTp with an E-value of 10–5 against the Database of Essential Genes (DEG). The five non-homolog proteins were classified as essential proteins and obligatory for the survival of the XDR Salmonella typhi strain and therefore, could be used as potential drug targets (Table 3). In principle, by targeting such proteins bacteria may survive but not be as virulent or many vital functions can be halted resulting in losing pathogenicity.
Virulent protein predictions
The virulence of shortlisted five hypothetical proteins was identified through the MP3 database (Gupta et al. 2014). Four proteins were classified as virulence proteins among the five proteins from earlier step and responsible for adverse effects in the host, whereas one was categorized as non-virulence protein (Table 4).
Subcellular localization prediction
It is important to know the subcellular localization for a better characterization of protein's function, molecular mechanism, chemical nature and organization (Yu et al. 2006). Protein localization is important to understand throughout the drug development process because it influences the design of novel drugs and vaccines. Cell membrane proteins, for example, are widely employed as vaccine targets, while cytoplasmic proteins are often used as therapeutic targets. Two tools i.e., PSORTb and CELLO2GO for sub-cellular localization identification were used for the shortlisted hypothetical proteins (Supplementary Data S1/Figure S2a/b).
Only one protein was identified as cytoplasmic membrane protein according to PSORTb results of subjected five proteins, whereas the other four were identified as present in unknown region. The PSORTb results of all the essential proteins is shown in Table 5. However, the CELLO2GO tool results are based on GO annotations that resulted in finding one protein as a cytoplasmic inner membrane protein, and three proteins were classified as cytoplasmic as well as inner membrane proteins. In addition, one protein was classified as periplasmic and inner membrane protein as shown in Table 6.
Phase 4: structure based studies
Structure based characterization of hypothetical proteins
Among shortlisted five proteins, only one protein was selected for further structure-based studies i.e. WP_000916613.1 based on its amino acid lengths, functional annotations, sub-cellular localization, virulence and stability comparison with other four hypothetical proteins (WP_000908466.1, WP_001681566.1, WP_100208313.1, and WP_001522276.1). These five proteins may be proposed as potential drug target due to their essential properties, non-homologous, virulent and resistant nature, and involvement in essential metabolic pathways. The overall classification of WP_000916613.1 protein as a potent drug target is shown in Table 7. The Fig. 6 highlights the stepwise filtering of the HPs as potential novel therapeutic targets for the current study.

Overview / Flowchart of the current study. Stepwise filtration of hypothetical proteins through comparative subtractive genomic analysis
Significance and metabolic pathway analysis of WP_000916613.1 protein
The WP_000916613.1 protein was classified through functional annotation studies as Endonucleases protein in nature that plays an essential role in DNA repair mechanism. It catalyzes the whole reaction as following:
2′-deoxyribonucleotide-(2′-deoxyribose 5′-phosphate)-2′-deoxyribonucleotide-DNA → 3′-end 2′-deoxyribonucleotide-(2,3-dehydro-2,3-deoxyribose 5′-phosphate)-DNA + a 5′-end 5′-monophospho-2′-deoxyribonucleoside-DNA + H+
The AP (Apurinic/apyrimidinic) endonuclease catalyzes the incision of DNA exclusively at AP sites for excision, repair synthesis and DNA ligation. The de-purination of DNA lesions results in a missing sugar along with the base (Fortini and Dogliotti 2007). The AP endonuclease recognizes that sugar and essentially cuts the DNA at this site and then allows for DNA repair to continue (Supplementary Data S1/Figure S3).
Structure modeling of drug target
The 3D structure of shortlisted HP was not available in PDB database. Therefore, the sequence was retrieved from the NCBI database to further study the structure and function through its accession ID: WP_000916613.1. Online BLAST was performed against the PDB database to find a possible template for WP_000916613.1. Among these, a template with higher quality (high query coverage and percent identity) was selected for modeling the structure (Uddin et al. 2017). The higher quality template was found from Haemophilus influenzae (O86237) having PDB ID: 1MWW with 30.5% sequence similarity. The structure of WP_000916613.1 was then modeled using the above-mentioned template (Supplementary Data S1/Figure S4).
Validation of the modeled structure
The modeled structure was verified through PSIPRED, PROCHECK server and ProFunc tool. The 2D structure of modeled protein was validated through PSIPRED. It validated the structure on the prediction of high number of helices and beta-sheet formation (as shown in Supplementary Data S1/Figure S5a) resulting in 6 helices and 3 beta sheets. The following steps nearly predicted the same position for the secondary structure as per the Homology Modeler.
The Ramachandran plot generated through PROCHECK validated the modeled structure by 88%. It showed about 88.2% residues in the most favorable region representing about 82 residues of the total sequence i.e. 104 residues. The additionally allowed region includes six residues making about 6.5% as shown in Supplementary Data S1/Figure S5b, turning it a good quality structure (McGuffin et al. 2000; Uddin et al. 2017).
The 3D modeled structure was also subjected to the validation of its function. The ProFunc tool verified the modeled structure according to its biochemical functional ability (Laskowski et al. 2005). It gives the modeled protein an ID of CR62. It also predicted the same secondary structure that was predicted by PSIPRED tool as shown in Fig. 7.

The validation of functional annotation through structure based analysis. The ProFunc analyzed the structure and predicted the same template and function as by other tools
Phylogenetic analysis of WP_000916613.1
The phylogenetic analysis was performed to identify the evolutionary relationship of WP_000916613.1 and its origin. The BLASTp of WP_000916613.1 protein was performed and related WP_000916613.1 proteins from various pathogens were retrieved. The phylogenetic analysis was performed for all of the WP_000916613.1 proteins from different organisms (Supplementary Data S1/Table S1, highlights the names and organism of phylogenetic tree proteins). It was observed that resistant pathogens are more frequent with WP_000916613.1 protein i.e. resistance to antibiotics either Unknown Resistivity, Single Drug Resistance or Multi Drug Resistance as shown in Fig. 8. Certainly, the expression and presence of WP_000916613.1 (Endonuclease like protein) may help pathogens to acquire its resistivity towards antibiotics.

Phylogenetic analysis of WP_000916613.1 protein (Green). Showing the evolutionary relation of VirB11 protein in other pathogens. The results the protein in Single Drug Resistance (Purple), MDR (Blue), XDR (Orange) pathogens and some are Unknown (Black)
Active site prediction
The prediction of protein’s active site is vital for various bioinformatics applications such as structure-based drug discovery and molecular docking studies. Accordingly, as during the modeling of the protein structures, there is a need to find interaction interface for the binding of the ligand. To do so, the DogSite Scorer tool was used. It predicted only four binding pockets for WP_000916613.1 protein whereas, first binding pocket was selected with high drug score (0.57) as shown in Supplementary Data S1/Figure S6. The actively found residues through DogSite Scorer within the binding cavity of WP_000916613.1 protein are represented in Table 8.
Protein–ligand interactions analysis
The protein–ligand interactions were analyzed by incorporating ligand identification, molecular docking and identified ligand–protein interactions through bioinformatics tools such as ProBis, AutoDock 4.2, PoseView, and Chimera.
Ligand identification
In drug target identification and drug research, the discovery of protein binding sites and their corresponding ligands plays an important role. Protein binding sites are a structurally and functionally crucial sites on the protein surface where various therapeutics interact to execute a desired activity. The ProBis server identified that the WP_000916613.1 protein share similar biding groove as F1-ATPase protein with PDB ID: 2JIZ (from Bos taurus) co crystalized with Resveratrol (STL) complex. Resveratrol was identified as a potent inhibitor to WP_000916613.1, IUPAC name as: 5-[(E)-2-(4-hydroxyphenyl) ethenyl] benzene-1,3-diol (Fig. 9a) with confidence score of 1.52 as the probable ligand. The resveratrol is a polyphenolic phytoalexin, the reported antibiotic (DB02709) extracted from plants with the help of stilbene synthase enzyme. It is used for the treatment of Herpes labialis infections (cold sores) (Sussman et al. 1998).

Ligand identification and molecular docking outcome. A Ligand Prediction. Ligand identified through ProBis for WP_000916613.1, i.e. PDB ID: STL, Commonly called as Resveratrol having IUPAC name of 5-[(E)-2-(4-hydroxyphenyl) ethenyl]benzene-1,3-diol, B Docked conformation of ligand with protein, and C Interaction of protein with ligand showing hydrogen and hydrophobic interaction
Molecular docking with AutoDock
The molecular docking analysis was performed with the identified ligand from the ProBis server and the modeled structure through AutoDock 4.2 tool. The ligand was docked followed by the parameters of 250 times Lamarckian GA settings resulting in 27,000 numbers of generations (Uddin and Azam 2019). The AutoDock results revealed different conformations and orientations of ligand binding at the active site of protein with diverse binding energies. The docking study resulted in the binding energy for resveratrol best docked conformation as − 8.46 kcal/mol. Figure 9b showed the high ranked conformation of docked ligand along with the structures.
Post docking analysis
The post docking and interaction analysis of resveratrol and HP was analyzed through chimera and Pose View tool. The results showed that resveratrol mediates two hydrophobic interactions and two hydrogen bonds with the receptor protein i.e. WP_000916613.1 (Fig. 9c). The two hydrogen bonds were formed by Trp82, and Met75 while neutral nonpolar amino acids Gln83, Leu74 were found as mediating two hydrophobic interactions.
Discussion
The genome annotation does not stop even after decoding the sequence. It continues to update as new information on protein homology and structure is discovered. It is observed that up to half of the S. typhi XDR H58 strain is comprised of the hypothetical proteins (Sivashankari and Shanmughavel 2006). The functional annotation and sub-cellular localization identification of these hypothetical proteins are of major importance that provides insights into the molecular function and to understand their interactions with the drug molecules and with other proteins inside the cell. This information is useful for the identification of new drugs against the disease (Pranavathiyani et al. 2020). Here, the main goal of the current study was to determine the function of the hypothetical proteins and eventually to prioritize potential drug targets among them against S. typhi XDR H58 (Ali et al. 2016; Sivashankari and Shanmughavel 2006).
In the current study, 535 hypothetical proteins were found in the XDR strain of Salmonella typhi. Therefore, the hypothetical proteins that are shortlisted through comparative genomic analysis were characterized using bioinformatics tools and various databases for homology similarity comparisons, domain identification, physicochemical characterization, cellular location, active site characterization, protein–protein interactions, and stability (Pranavathiyani et al. 2020). The strategy used in the current study to annotate functions of hypothetical proteins can be useful for designing experimental approaches geared towards the evolution of the exact function of the corresponding protein (Klemm et al. 2018).
The comparative proteo-genomic approach was applied to these 535 hypothetical proteins. About 351 proteins were shortlisted from these 535 hypothetical proteins as uniquely present in the XDR strain (i.e. comparison with MDR and NR strains of Salmonella typhi). Moreover, functional annotation of these 351 hypothetical proteins was performed through various computational tool i.e. Argot2, PFP, ProtoNet, and SMART. The functional prediction ability of these tools was further validated through ROC analysis with the submission of 100 known functional proteins from the XDR strain of Salmonella typhi. The overall accuracy of these tools was found to be 91% making five hypothetical proteins functional prediction more effectual. The physicochemical properties for these shortlisted proteins were estimated through ProtParam server that helped to understand the nature and type of proteins. Similar functional analysis was performed in Human Adenovirus by Naveed et al. (Naveed et al. 2017) and in Exiguobacterium antarcticum B7 by Klemm et al. (Klemm et al. 2018). Consequently, the function to hypothetical proteins from H58 strain was assigned with a high confidence (Supplementary data S2) with specific characterization into cellular localization and physico-chemical properties. Furthermore, subtractive genomic analysis was performed to these 351 hypothetical proteins. Out of these, 350 hypothetical proteins were characterized as non-homolog proteins (in comparison to the human proteome). Consequently, five proteins were shortlisted as essential, drug target like, virulent non-homologous proteins and found only in the XDR strain of Salmonella typhi. Besides identification of function of hypothetical proteins, the main interest was to identify the potential drug targets in XDR H58 strains. From the above study, only one protein WP_000916613.1 was found to be fully characterized and was selected for further structure-based studies. Conclusively, secondary structure prediction and 3D modeling further provided insights into the spatial arrangement of the amino acids in the proteins to find out the most probable binding sites for the drugs.
Certainly, the current study helped in the prediction of the function of hypothetical proteins that may be proposed as potential drug targets. The predicted results can be further validated through experimental studies. Therefore, it is hoped that the information of hypothetical proteins from S. typhi from the current study will be helpful for further in-vitro analysis of Salmonella typhi disease.
Conclusion
The proteins are versatile macromolecules that play a crucial role in the biological processes. The identification of protein function is fundamental for the understanding of these processes (Jamilah et al. 2020). An in silico subtractive genomics based approach was applied in this study to predict the function of hypothetical proteins from the XDR H58 strain of Salmonella typhi (Klemm et al. 2018). These hypothetical proteins may serve as potential therapeutic targets. While this contributes to the current understanding of the S. typhi XDR strain, still there is more to be explored in this field. More homologous proteins and structural information are needed in public repositories to fully evaluate some hypothetical proteins. For appropriate annotation and maintenance of entire genome, this process should repeat necessarily at regular intervals. The automation of this process would help ensure up-to-date databases. Until then, the data describing what is currently available for XDR hypothetical proteins will contribute to the scientific understanding of S. typhi aiding in the discovery of therapeutic targets.
Data availability
All the data are included in the manuscript and as supplementary files.
References
Akram J, Khan AS, Khan HA, Gilani SA, Akram SJ, Ahmad FJ, Mehboob R (2020) Extensively drug-resistant (XDR) typhoid: evolution, prevention, and its management. Biomed Res Int 2020:6432580
Ali M, Ahsan Z, Amin M, Latif S, Ayyaz A, Ayyaz M (2016) ID-Viewer: a visual analytics architecture for infectious diseases surveillance and response management in Pakistan. Public Health 134:72–85
Bhairamadgi RN, Katti AKS (2013) In-silico identification and sequence annotations of potential vaccine candidate in neisseria gonorrhoeae. Int J Adv Biotechnol Res 4:404–414
Consortium U (2015) UniProt: a hub for protein information. Nucleic Acids Res 43:D204–D212
da Costa WLO, Araújo CLDA, Dias LM, Pereira LCDS, Alves JTC, Araujo FA, Folador EL, Henriques I, Silva A, Folador ARC (2018) Functional annotation of hypothetical proteins from the Exiguobacterium antarcticum strain B7 reveals proteins involved in adaptation to extreme environments, including high arsenic resistance. PLoS ONE 13:e0198965
Dannheim H, Riedel T, Neumann-Schaal M, Bunk B, Schober I, Spröer C, Chibani CM, Gronow S, Liesegang H, Overmann J (2017) Manual curation and reannotation of the genomes of Clostridium difficile 630Δerm and C. difficile 630. J Med Microbiol 66:286–293
Falda M, Toppo S, Pescarolo A, Lavezzo E, Di Camillo B, Facchinetti A, Cilia E, Velasco R, Fontana P (2012) Argot2: a large scale function prediction tool relying on semantic similarity of weighted Gene Ontology terms. BMC Bioinform 13:1–9
Feasey NA, Gaskell K, Wong V, Msefula C, Selemani G, Kumwenda S, Allain TJ, Mallewa J, Kennedy N, Bennett A (2015) Rapid emergence of multidrug resistant, H58-lineage Salmonella typhi in Blantyre. Malawi. Plos Negl Trop Dis 9:e0003748
Fenoll A, Granizo J, Aguilar L, Giménez M, Aragoneses-Fenoll L, Hanquet G, Casal J, Tarragó D (2009) Temporal trends of invasive Streptococcus pneumoniae serotypes and antimicrobial resistance patterns in Spain from 1979 to 2007. J Clin Microbiol 47:1012–1020
Fiser A, Šali A (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491
Fortini P, Dogliotti E (2007) Base damage and single-strand break repair: mechanisms and functional significance of short-and long-patch repair subpathways. DNA Repair 6:398–409
Garg VK, Avashthi H, Tiwari A, Jain PA, Ramkete PW, Kayastha AM, Singh VK (2016) MFPPI–multi FASTA ProtParam interface. Bioinformation 12:74
Goksuluk D, Korkmaz S, Zararsiz G, Karaagaoglu AE (2016) easyROC: an interactive web-tool for ROC curve analysis using R language environment. The R Journal 8:213
Grant JR, Stothard P (2008) The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res 36:W181–W184
Gupta A, Kapil R, Dhakan DB, Sharma VK (2014) MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLoS ONE 9:e93907
Hawkins T, Chitale M, Luban S, Kihara D (2009) PFP: automated prediction of gene ontology functional annotations with confidence scores using protein sequence data. Proteins 74:566–582
Islam MS, Shahik SM, Sohel M, Patwary NI, Hasan MA (2015) In silico structural and functional annotation of hypothetical proteins of Vibrio cholerae O139. Genomics Inform 13:53
Jamilah J, Hatta M, Natzir R, Umar F, Sjahril R, Agus R, Junita A, Dwiyanti R, Primaguna M, Sabir M (2020) Analysis of existence of multidrug-resistant H58 gene in Salmonella enterica serovar Typhi isolated from typhoid fever patients in Makassar, Indonesia. New Microbes New Infect 38:100793
Klemm EJ, Shakoor S, Page AJ, Qamar FN, Judge K, Saeed DK, Wong VK, Dallman TJ, Nair S, Baker S (2018) Emergence of an extensively drug-resistant Salmonella enterica serovar Typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation cephalosporins. Mbio 9:e00105-00118
Konc J, Janežič D (2010) ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 26:1160–1168
Laskowski RA, Watson JD, Thornton JM (2005) ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 33:W89–W93
Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16:404–405
Naveed M, Tehreem S, Usman M, Chaudhry Z, Abbas G (2017) Structural and functional annotation of hypothetical proteins of human adenovirus: prioritizing the novel drug targets. BMC Res Notes 10:1–6
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pranavathiyani G, Prava J, Rajeev AC, Pan A (2020) Novel target exploration from hypothetical proteins of Klebsiella pneumoniae MGH 78578 reveals a protein involved in host-pathogen interaction. Front Cell Infect Microbiol 10:109
Pruitt KD, Tatusova T, Maglott DR (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33:D501–D504
Rasheed F, Saeed M, Alikhan N-F, Baker D, Khurshid M, Ainsworth EV, Turner AK, Imran AA, Rasool MH, Saqalein M, Nisar MA, Fayyaz ur Rehman M, Wain J, Yasir M, Langridge GC, Ikram A (2020) Emergence of resistance to fluoroquinolones and third-generation cephalosporins in Salmonella typhi in Lahore, Pakistan. Microorganisms. https://doi.org/10.3390/microorganisms8091336
Sah R, Donovan S, Seth-Smith HM, Bloemberg G, Wüthrich D, Stephan R, Kataria S, Kumar M, Singla S, Deswal V (2020) A novel lineage of ceftriaxone-resistant Salmonella typhi from India that is closely related to XDR S. Typhi found in Pakistan. Clin Infect Dis 71:1327–1330
Sasson O, Vaaknin A, Fleischer H, Portugaly E, Bilu Y, Linial N, Linial M (2003) ProtoNet: hierarchical classification of the protein space. Nucleic Acids Res 31:348–352
Schultz J, Milpetz F, Bork P, Ponting CP (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 95:5857–5864
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Sivashankari S, Shanmughavel P (2006) Functional annotation of hypothetical proteins—a review. Bioinformation 1:335
Stierand K, Rarey M (2010) PoseView–molecular interaction patterns at a glance. J Cheminformatics 2:1–1
Sussman JL, Lin D, Jiang J, Manning NO, Prilusky J, Ritter O, Abola EE (1998) Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr Sect D 54:1078–1084
Thanh DP, Karkey A, Dongol S, Thi NH, Thompson CN, Rabaa MA, Arjyal A, Holt KE, Wong V, Thieu NTV (2016) A novel ciprofloxacin-resistant subclade of H58 Salmonella Typhi is associated with fluoroquinolone treatment failure. Elife 5:e14003
Uddin R, Azam SS (2019) Identification of glucosyl-3-phosphoglycerate phosphatase as a novel drug target against resistant strain of Mycobacterium tuberculosis (XDR1219) by using comparative metabolic pathway approach. Comput Biol Chem 79:91–102
Uddin R, Saeed K (2014) Identification and characterization of potential drug targets by subtractive genome analyses of methicillin resistant Staphylococcus aureus. Comput Biol Chem 48:55–63
Uddin R, Tariq SS, Azam SS, Wadood A, Moin ST (2017) Identification of histone deacetylase (HDAC) as a drug target against MRSA via interolog method of protein-protein interaction prediction. Eur J Pharm Sci 106:198–211
Uddin R, Siraj B, Rashid M, Khan A, Ahsan Halim S, Al-Harrasi A (2020) Genome subtraction and comparison for the identification of novel drug targets against Mycobacterium avium subsp. hominissuis. Pathogens 9:368
Varma PBS, Adimulam YB, Kodukula S (2015) In silico functional annotation of a hypothetical protein from Staphylococcus aureus. J Infect Public Health 8:526–532
Volkamer A, Kuhn D, Rippmann F, Rarey M (2012) DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 28:2074–2075
Wadood A, Jamal A, Riaz M, Khan A, Uddin R, Jelani M, Azam SS (2018) Subtractive genome analysis for in silico identification and characterization of novel drug targets in Streptococcus pneumonia strain JJA. Microb Pathog 115:194–198
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082
Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64:643–651
Yu C-S, Cheng C-W, Su W-C, Chang K-C, Huang S-W, Hwang J-K, Lu C-H (2014) CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 9:e99368
Zhang R, Ou HY, Zhang CT (2004) DEG: a database of essential genes. Nucleic Acids Res 32:D271–D272
Supplementary information
Supplementary Data S1—Phylogenetic proteins detailed and Supplementary figures.
Supplementary Data S2—Functional Annotation. Elaborates the function annotation of shortlisted HPs through PFP, ArGot2, ProtoNet, and SMART tool.
Supplementary Data S3—Known Protein Functional Annotation. Validation of functional annotation of tools used for HPs through 100 known proteins. Table comprises of Protein names and score generated through these tools.
Supplementary Data S4—Physico-chemical Properties. Physico-chemical properties calculated for shortlisted protein through ProtParam tool.
Funding
The authors would like to acknowledge the Pakistan Science Foundation and International Foundation for Science (IFS) for providing the financial support.
Author information
Authors and Affiliations
Contributions
KK and RU conceived and designed the study. KK performed data collection and analysis, and contributed to drafting of the manuscript. RU provided technical and material support and supervised the study. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interests associated with the manuscript.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khan, K., Uddin, R. Integrated bioinformatics based subtractive genomics approach to decipher the therapeutic function of hypothetical proteins from Salmonella typhi XDR H-58 strain. Biotechnol Lett 44, 279–298 (2022). https://doi.org/10.1007/s10529-021-03219-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-021-03219-6