
Abstract
Objectives
Enhancement of the potential ability of biomacromolecules to cross cell membranes is a critical step for development of effective therapeutic vaccine especially DNA vaccine against human immunodeficiency virus-1 (HIV-1) infection. The supercharged proteins were known as powerful weapons for delivery of different types of cargoes such as DNA and protein. Hence, we applied B1 protein with + 43 net charges obtained from a single frameshift in the gene encoding enhanced green fluorescent protein (eGFP) for delivery of two multi-epitope DNA constructs (nef-vpu-gp160-p24 and nef-vif-gp160-p24) in vitro and in vivo for the first time. For this purpose, B1 protein was generated in bacterial expression system under native conditions, and used to interact with both DNA constructs.
Results
Our data indicated that B1 protein (~ 27 kDa) was able to form a stable nanoparticle (~ 80–110 nm) with both DNA constructs at nitrogen: phosphate (N: P) ratio of 1:1. Moreover, the transfection efficiency of B1 protein for DNA delivery into HEK-293T cell line indicated that the cellular uptake of nef-vif-gp160-p24 DNA/ B1 and nef-vpu-gp160-p24 DNA/ B1 nanoparticles was about 32–35% with lower intensity as compared to TurboFect commercial reagent. On the other hand, immunization of BALB/c mice with different modalities demonstrated that B1 protein could enhance the levels of antibody, IFN-gamma and Granzyme B eliciting potent and strong Th1-directed cellular immunity.
Conclusion
Generally, our findings showed the potency of B1 protein as a promising gene delivery system to improve an effective therapeutic vaccine against HIV-1 infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the biggest challenges in vaccine development is their inability to stimulate efficient immune responses in the targeted host. This drawback led to improvement of various approaches for DNA and protein/polypeptide delivery into cells (Kardani et al. 2019a, b). Some of these approaches are electroporation (Sardesai and Weiner 2011), nebulization (Rajapaksa et al. 2014), liposomes (Schwendener 2014), cationic polymers (Bolhassani et al. 2014), polyethyleneimine (PEI) (Ma and Yang 2010), gold nanoparticles (Dings et al. 2018), carbon nanotubes (Hasnain and Nayak 2019), and cell penetrating peptides (Bolhassani et al. 2015). Among them, cell-penetrating peptides could deliver different types of cargoes including DNA, proteins, liposomes, and inorganic particles in a safe way, and enhance the induction of both humoral and cellular immune responses (Kardani et al. 2019a, b; Bolhassani et al. 2015; Motevalli et al. 2018). Recently, various unnatural CPPs were prepared such as amphipathic peptides (e.g., Cady and Pep) (Kurzawa et al. 2010), highly positive charge proteins (e.g., supercharged green fluorescent protein: scGFP) (Motevalli et al. 2018; Shahbazi et al. 2018), and polyarginine peptides (e.g., HR9) (Crosio et al. 2019) for nucleic acid, protein or polypeptide delivery. Their cellular uptake is through direct penetration or endocytic pathways (Kardani et al. 2019a, b). It has proved by numerous studies that positively charge density of carriers enhanced their endosomal escape (McNaughton et al. 2009). For example, the scGFP has a theoretical net charge of + 36 (+ 36 GFP) by replacement of anionic amino acids in enhanced green fluorescent protein (eGFP) with cationic residues (e.g., Arg and Lys) (Motevalli et al. 2018; Brock et al. 2018). It is interesting to know that the supercharged CPPs are available in nature, as well. Several supercharged proteins exist in the capsids of different viruses including yellow fever virus (YFC) with a net charge of + 52, Dengue virus (DENV) with a net charge of + 42, and West Nile virus (WNV) with a net charge of + 46 (Freire et al. 2013; Freire et al. 2015b, a). Simeon et al. discovered a novel supercharged CPP known as B1 protein which has 244 residues in length and also a net charge of + 43 at neutral pH originated from a frameshifted eGFP. It was reported that B1 protein could deliver non-covalently nucleic acid cargoes and genetically fused proteins (Simeon and Chen 2013; Simeon et al. 2013).
Acquired immune deficiency syndrome (AIDS) caused by human immunodeficiency virus type- 1 (HIV-1) is a problem of public health (Cluck and Underwood 2018). The genome of HIV-1 comprises of structural (env, gag and pol), regulatory (rev and tat), and accessory (nef, vpu, vif and vpr) genes. The HIV-1 genes encode 16 viral proteins in total (Li and De Clercq 2016). The absence of a potent vaccine against hypervariable HIV-1 led to novel design of regimens and approaches. One of these strategies is the use of immune-informatics tools to find highly conserved and most immunogenic T-cell epitopes, and develop an effective multi-epitope vaccine (MEV) (Cluck and Underwood 2018; Li and De Clercq 2016; Kardani et al. 2019a, b). In our previous study, two multi-epitope-based vaccine candidates consisting various immunogenic T-cell epitopes of HIV-1 proteins (Nef-Vif-Gp160-P24 and Nef-Vpu-Gp160-P24) were designed against both human and mouse MHC molecule alleles (Kardani et al. 2019a, b). In the current report, the supercharged B1 CPP was generated in bacterial expression system, and its ability was evaluated to deliver both multi-epitope-based vaccine candidates in vitro and in vivo and enhance immune responses for the first time. Herein, the potency of different immunization modalities including DNA prime/DNA boost, DNA prime/multi-epitope peptide boost, and multi-epitope peptide prime/multi-epitope peptide boost were evaluated and compared to effectively induce cellular immune responses.
Materials and methods
In vitro experiments
Figure 1 shows the overall process of in vitro experiments.

The overall process of in vitro studies
Construction of the recombinant plasmids
At first, the pUC57-B1 construct was synthesized by Biomatik Corporation (Canada). Then, the B1 gene fragment (Simeon and Chen 2013) was subcloned into eukaryotic (pEGFP-N1: 4733 bp; CMV promoter), and prokaryotic (pET-24a: 5310 bp; T7 promoter) vectors.
Cloning of the B1 gene into pEGFP-N1 vector
To ensure the green fluorescence feature of B1 protein, the eGFP gene of pEGFP-N1 was replaced with the B1 gene. For this purpose, the pEGFP-N1 was digested by NheI/NotI restriction enzymes (Thermo Fisher Scientific), and then B1 gene fragment was inserted into NheI/NotI sites of pEGFP-N1 vector (without GFP) with T4 DNA ligase (Thermo Fisher Scientific) as shown in Fig. 2. The Escherichia coli (E. coli) DH5α strain was transformed with the recombinant plasmid. The recombinant plasmid was purified by ion-exchange chromatography using DNA extraction mini kit (FAVORGEN, Taiwan). The concentration and purity of the DNA plasmid (named as pEGFP-B1) was determined by NanoDrop spectrophotometry.

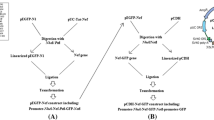
The schematic process of B1 gene cloning in a pET-24a(+), and b pEGFP-N1: a the B1 fragment was digested from pUC57 using BamHI and HindIII, and then ligated into pET-24a(+) vector; b In order to clone B1 gene into pEGFP-N1, at first eGFP gene was excluded from pEGFP-N1 vector with NheI and NotI restriction enzymes. Next, B1 gene fragment was digested with the same restriction enzymes (NheI and NotI). Finally, B1 gene was ligated into pEGFP-N1 vector
Cloning of the B1 gene into pET-24a (+) vector
The pET-24a (+) was used to express the recombinant B1 protein. The B1 gene was digested by BamHI/HindIII restriction enzymes (Thermo Fisher Scientific), and then subcloned into the BamHI/HindIII sites of the pET-24a (+) prokaryotic expression vector (Invitrogen, USA) using T4 DNA ligase (Thermo Fisher Scientific). The competent E. coli DH5α strain was transformed with the recombinant pET-24a (+)-B1. Finally, the presence of the B1 gene was confirmed by digestion with the restriction enzymes on agarose gel electrophoresis (Fig. 2).
Expression of the B1 protein in bacterial expression system
Two different E. coli strains such as Rosetta (DE3) and BL21 (DE3) were transformed with the recombinant pET-24a (+)-B1 vector using heat shock. Then, protein induction was done by isopropyl-β-d-thiogalactopyransoide (IPTG, 1 mM, SinaClon bioscience Co, Iran) at 37 °C and different times (2, 3, 4 and 16 h) with shaking at 150 rpm. In the next step, the lysate of bacteria expressing B1 protein was collected and analyzed by Poly Acrylamide Gel Electrophoresis on 12% (W/V) sodium dodecyl sulfate (SDS-PAGE). The gel was stained using Coomassie brilliant blue to observe the protein band. In the next step, the expression of B1 protein was confirmed by western blot analysis using an anti-His-tag antibody (Abcam, USA).
Purification of the B1 protein
The B1 protein was purified by nickel-nitrilotriacetic acid (Ni–NTA)-agarose column (Qiagen, Germany) under native conditions (i.e., 300 mM imidazole buffer, pH 8) using 6xHis-tag according to manufacturer’s instructions. Then, the purified B1 protein was dialyzed in phosphate-buffered saline (PBS) 1X using a 10 kDa dialysis membrane (Thermo Scientific). Finally, the concentration of protein was determined by Bradford protein assay kit (Sigma, Germany) and NanoDrop spectrophotometer at 280 nm. The purified B1 protein was validated by western blot analysis using an anti-His-tag antibody (Abcam, USA).
Preparation of the B1/DNA complexes
The pEGFP-nef-vpu-gp160-p24 and pEGFP-nef-vif-gp160-p24 constructs were previously prepared by our group (Kardani et al. 2020). In order to form B1/DNA complexes, 2 µg of DNA constructs were mixed with B1 protein at different N: P ratios of 0.5, 0.75, 1 and 1.5 in PBS 1X, and incubated for 30 min at room temperature. To detect the mobility of B1/DNA complexes, gel electrophoresis on 1% agarose was used.
Physicochemical features of the B1/DNA complexes
The charge of nanoparticles, and their morphology and size at N: P ratio of 1:1 was determined by Zetasizer Nano ZS (Malvern Instruments, UK) at 25 °C, and scanning electron microscope (SEM; KYKY-EM3200 model, China), respectively.
Stability and protection assay of the B1/DNA complexes
To recognize the stability of pEGFP-N1-nef-vpu-gp160-p24/B1 and pEGFP-N1-nef-vif-gp160-p24/B1 nanoparticles against DNA nucleases, DNase I was added to the complexes with an N: P ratio of 1:1 and then, the mixture was incubated for 60 min at 37 °C. Finally, the stop solution (1% SDS, 200 mM sodium chloride, and 20 mM EDTA) was added to the mixture. In addition, to determine the serum stability, the B1/pEGFP-nef-vpu-gp160-p24 and B1/pEGFP-nef-vif-gp160-p24 nanoparticles at N: P ratio of 1:1 were subjected to 10% serum and incubated at 37 °C for 5 h (h). Next, 10% SDS was added to the mixture for 2 h to dissociate the DNA plasmids. Finally, 1% agarose gel was employed to analyze the intact DNA plasmids.
Cell culture
Human embryonic kidney cell (HEK-293 T; ATCC: CRL-3216™, Pasteur Institute of Iran) was cultured in RPMI 1640 medium (Sigma, Germany), supplemented with 10% fetal bovine serum (FBS, Gibco, Germany), and 1% Gentamicin solution under 5% CO2, 37 °C, and 85% relative humidity conditions.
MTT cell proliferation and cytotoxicity assay
To evaluate the cytotoxicity of B1 protein, B1/pEGFP-nef-vpu-gp160-p24 nanoparticles, and B1/pEGFP-nef-vif-gp160-p24 nanoparticles, the MTT proliferation assay was performed (Sadeghian et al. 2012). Hence, the HEK-293 T cells (10,000/well) were cultured onto 96-well culture plates. After obtaining about 80–85% confluency, the medium was replaced with fresh RPMI 1640. Then, the cells were treated with each compound for 48 h. Next, medium was removed, and the 3-(4, 5- dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT yellow dye, 5 mg/ml, Sigma, Germany) was added to the cells, and incubated at 37 °C for 3 h in a humidified 5% CO2. After the incubation period, the medium was removed, and dimethyl sulfoxide (DMSO) was added to dissolve purple formazan crystals. The absorbance was measured by ELISA reader (Labsystems Multiskan MS 352 Microplate Reader) at 570 nm. The cells treated with 70% ethanol, and untreated cells were considered as positive and negative controls, respectively. The MTT proliferation assay was done in triplicate.
The B1-mediated delivery of DNA constructs
The HEK-293T cells were cultured onto 24-well plates at a density of 5 × 104 cells/well in RPMI 1640 containing 10% FBS, and incubated overnight. After growth of the HEK-293T cells to 85% confluency, the medium was replaced by serum-free medium containing the compounds such as B1 protein, B1/pEGFP-nef-vpu-gp160-p24 nanoparticle, B1/pEGFP-nef-vif-gp160-p24 nanoparticle, pEGFP-N1/TurboFect, pEGFP-B1/TurboFect, pEGFP-nef-vpu-gp160-p24/TurboFect and pEGFP-nef-vif-gp160-p24/TurboFect, and incubated at 37˚C and 5% CO2 atmosphere for 6 h. After incubation time, the cells were supplemented with fresh completed RPMI 1640 without removal of the complexes, and incubated for 48 h at 37 °C, 5% CO2 and 85% humidity. The cells treated with TurboFect/pEGFP-N1, and untreated cells were used as positive and negative controls, respectively. Finally, the transfection efficiency was determined by fluorescent microscopy, flow cytometry, and western blot analysis using an anti-His-tag antibody (Abcam, USA) for detection B1 protein and B1/DNA nanoparticles as well as anti-GFP antibody (Abcam, USA) for detection of wild type GFP at 48 h after transfection. For flow cytometry and western blotting, the cells were treated with trypsin–EDTA, harvested, washed three times, and resuspended in PBS 1X.
Generation of multi-epitope peptides in E.coli
The recombinant (r) Nef-Vpu-Gp160-P24 and Nef-Vif-Gp160-P24 multi-epitope peptides were expressed in pET/Rosetta (DE3) system and purified by affinity chromatography under denaturing conditions as reported in our previous study (Kardani et al. 2020). The sequences of both multi-epitope peptides were shown in Fig. 3.

The overall process of in vivo studies: The timeline illustrates the immunization days. The first immunization was subcutaneously done at day 0. The first and second boosters were subcutaneously performed at days 14 and 28, respectively. The immunized mice were bled from retro-orbital to determine the humoral immune responses at day 55. At the end, three mice were sacrificed randomly for each group at day 58. Then, spleens were collected and the red blood cell depleted pooled splenocytes were cultured to measure cellular responses and Granzyme B secretion
In vivo experiments
Figure 4 indicates the overall process of in vivo experiments.

The schematic model of the designed multi-epitope peptide constructs: a Nef-Vif-Gp160-P24; b Nef-Vpu-Gp160-P24
Mice immunization
Six-to-eight-week-old BALB/c female mice from the Pasteur Institute of Iran were used for immunization. Twelve groups of four mice were immunized subcutaneously at the footpad three times with two-week intervals. The immunization program was indicated in Table 1. In addition, all mice were maintained under pathogen-free conditions, and the whole in vivo experiments procedures was approved by protocols and care of laboratory animals at Pasteur Institute of Iran (ethical code:1070). Montanide ISA720 was used as an adjuvant for multi-epitope peptides.
Antibody assay
Four weeks after the final injection, the mouse groups were bled from retroorbital (after anesthesia using intraperitoneal injection of Ketamine (87.5 mg/kg)/Xylazine (12.5 mg/kg) cocktail: 0.1 ml/20 g mouse) and the serum samples were collected from each group (n = 4) and the pooled sera were stored at − 20 °C. The secretion of total immunoglobulin G (IgG) and IgG1, IgG2a and IgG2b isotypes (1:10,000 v/v, Sigma, Germany) in the sera were measured using indirect ELISA. The rNef-Vif-Gp160-P24 (2.5 µg/ml) and rNef-Vpu-Gp160-P24 (2.5 µg/ml) multi-epitope peptides (diluted in PBS 1X) were used as coated antigens. The 3, 3′, 5, 5′-tetramethylbenzidine (TMB, Sigma, Germany) was employed as a substrate, and the absorbance was measured at 450 nm (Kardani et al. 2020).
Cellular assay
Four weeks after the last injection, all mice from each group (n = 4) were sacrificed and the spleens were removed. The red blood cell-depleted splenocytes (2 × 106 cells per well) were cultured in 48-well plates (Costar, Cambridge, MA) for 72 h in the presence of 2.5 µg/ml of rNef-Vpu-Gp160-P24 and rNef-Vif-Gp160-P24 multi-epitope peptides. RPMI 5% and 5 µg/ml concanavalin A (Con A) were used as negative and positive controls in complete RPMI culture medium. The supernatant was harvested for further evaluation and frozen at -70 °C. The sandwich-based ELISA system (Mabtech, Sweden) was applied to assess the levels of IL-5 and IFN-γ cytokines according to the manufacturer’s handbook. All data were represented as mean ± SD for each set of samples. The lower detection limit was 4 pg/ml for both cytokines.
Measurement of Granzyme B (GrB) secretion
SP2/0 target cells (T, mouse hybridoma cells, provided from the cell bank at Pasteur Institute of Iran) were seeded in triplicate into U-bottomed, 96-well plates (2 × 104 cells/well) incubated with rNef-Vpu-Gp160-P24 and rNef-Vif-Gp160-P24 antigens (~ 2.5 µg/ml) for 24 h. The splenocytes (E: Effector cells) prepared in the last section were counted, and added to the target cells at E: T ratio of 100:1 (maximal release of Granzyme B) in complete RPMI culture medium. After 6 h of incubation, the microplates were centrifuged at 4 °C for 5 min at 250×g. Then, the supernatants were harvested and the ELISA kit (eBioscience, USA) was used to measure the concentration of Granzyme B (GrB) according to the manufacturer’s instruction.
Statistical analysis
Statistical analysis was performed using Prism 5.0 software (GraphPad, San Diego, CA). One-way ANOVA and Student’s t-test were used to recognize the differences in the levels of humoral and cellular responses between the test and control groups. For all analyses, p-value < 0.05 was considered statistically significant. Data are represented as mean ± standard deviation (SD).
Results
In vitro experiments
Construction, expression and purification of B1 protein
The B1 gene was correctly subcloned in mammalian and bacterial expression vectors. The existence of the B1 gene in pET-24a and pEGFP-N1 was validated using double digestion as clear bands of ~ 757 and ~ 796 bp, respectively (Fig. 5). Moreover, the recombinant B1 protein was generated in pET-24a/Rosetta expression system under 1 mM IPTG, overnight period and 37 °C conditions as a clear band of ~ 27 kDa on 12.5% SDS-PAGE. Then, purification of the B1 protein was done using affinity chromatography under native conditions (Fig. 6). Finally, the purified B1 protein was detected by anti-His-tag antibody using western blot analysis and its concentration was obtained about 3–4 mg/ml.

Confirmation of B1 gene fragment cloned in a pET-24a (+), and b pEGFP-N1. a Lane 1: undigested pET-24a (+)-B1, Lane 2: double digested pET-24a (+) and B1 gene (~ 757 bp) using BamHI/HindIII restriction enzymes; b Lane 1: double digested pEGFP-N1 and B1 gene (~ 796 bp) using NheI/NotI restriction enzymes; MW is molecular weight marker (1 kb, Fermentas)

Expression and purification of B1 protein in E. coli Rosetta expression system: Lane 1: Before induction, Lane 2: 3 h post-induction, Lane 3: 4 h post-induction, Lane 4: 16–18 h post-induction, Lane 5: the purified B1 protein using affinity chromatography under native conditions. The purified B1 protein migrated as a dominant band of ~ 27 kDa in SDS-PAGE; MW is molecular weight marker (10–180 kDa, Fermentas)
Characterization of the B1/DNA nanoparticles
The formation of B1/DNA nanoparticles and their stability against DNase and serum were confirmed at N/P ratio of 1:1 through 1% agarose gel electrophoresis. Indeed, the nanoparticles were generated by interaction of the cationic B1 protein with the negatively charged pEGFP-nef-vif-gp160-p24 and pEGFP-nef-vpu-gp160-p24 constructs. The plasmid DNA in complexes remained intact. The zeta potential and SEM results of pEGFP-nef-vif-gp160-p24/B1 and pEGFP-nef-vpu-gp160-p24/B1 nanoparticles were determined at a certain N/P ratio of 1:1. The pEGFP-nef-vif-gp160-p24/B1 and pEGFP-nef-vpu-gp160-p24/B1 nanoparticles showed the positive charges of + 18.2 and + 21 mV, respectively; whereas the zeta potential of pEGFP-nef-vif-gp160-p24 and pEGFP-nef-vpu-gp160-p24 was about -21.7 and -28.8 mV, respectively. On the other hand, the pEGFP-nef-vif-gp160-p24/B1 and pEGFP-nef-vpu-gp160-p24/B1 nanoparticles indicated a diameter of ~ 79–82 nm and ~ 99–105 nm, respectively. These nanoparticles with a positive charge and a small diameter can penetrate efficiently through the cell membrane.
Cytotoxicity assay
According to the cell viability results, the B1/pEGFP-N1-nef-vif-gp160-p24 and B1/pEGFP-N1-nef-vpu-gp160-p24 nanoparticles at N/P ratio of 1:1 as well as the used B1 protein did not induce any visible cytotoxicity effects in HEK-293 T cell lines after 48 h incubation compared to untreated cells (cell viability: 91–96%). The cell viability of untreated cells was ~ 95–99%, as well (data not shown). Therefore, these nanoparticles were not toxic at the used doses in vitro and in vivo.
The B1-mediated delivery of DNA constructs
The B1 protein could mediate internalization of pEGFP-nef-vif-gp160-p24 and pEGFP-nef-vpu-gp160-p24 efficiently into HEK-293T cell lines as shown in Fig. 7. The flow cytometry results were indicated by the percentage of protein expression using B1 reporter marker. The results showed that the mean of cellular uptake for B1 protein, B1/pEGFP-nef-vif-gp160-p24 nanoparticle, and B1/pEGFP-nef-vpu-gp160-p24 nanoparticle was ~ 60.19%, ~ 35.64% and ~ 32.63%, respectively. It was previously observed that the transfection efficacy was ~ 53.72% and ~ 66.85% for the TurboFect/pEGFP-nef-vpu-gp160-p24 and TurboFect/pEGFP-nef-vif-gp160-p24 complexes, respectively (Kardani et al. 2019a, b). In addition, the transfection efficiency of TurboFect/pEGFP-N1 and TurboFect/pEGFP-B1 was ~ 95.69% and ~ 68.55%, respectively. Regarding these data, B1 protein could penetrate efficiently and also enhance DNA delivery in the cells. On the other hand, B1 protein was expressed successfully instead of eGFP using TurboFect transfection reagent. Western blot analysis validated the permeability of pEGFP-N1/TurboFect, pEGFP-B1/TurboFect, B1 protein, pEGFP-nef-vif-gp160-p24/B1, and pEGFP-nef-vpu-gp160-p24/B1 using an anti-His-tag antibody (Fig. 7). Our data indicated a single band of ~ 32 kDa in the cells treated with the pEGFP-nef-vif-gp160-p24/B1 and pEGFP-nef-vpu-gp160-p24/B1 nanoparticles, and a single band of ~ 27 kDa for eGFP and B1 proteins as compared to untransfected cells.

The transfection efficiency of multi-epitope DNA constructs delivered by B1 protein as compared to TurboFect transfection reagent in HEK-293T cells using flow cytometry: a untransfected HEK-293T cells; b cells transfected by pEGFP-N1/TurboFect; c cells transfected by pEGFP-B1/TurboFect; d cells transfected by B1 protein; e cells transfected by B1/ pEGFP-nef-vif-gp160-p24; f cells transfected by B1/pEGFP-nef-vpu-gp160-p24; b Western blot analysis of the transfected HEK-293T cells: Lane (1) Untransfected cells as negative control, Lane (2) cells transfected by pEGFP-N1/TurboFect, Lane (3) cells transfected by pEGFP-B1/TurboFect, Lane (4) cells transfected by B1 protein, Lane (5) cells transfected by B1/pEGFP-nef-vif-gp160-p24, and Lane (6) cells transfected by B1/pEGFP-nef-vpu-gp160-p24; A dominant band of ~ 32 kDa was observed for delivery of pEGFP-N1-nef-vif-gp160-p24 and pEGFP-N1-nef-vpu-gp160-p24 using B1 protein. MW is molecular weight marker (10–180 kDa, Fermentas)
In vivo experiments
Antibody response in mice
The anti-Nef-Vif-Gp160-P24/Nef-Vpu-Gp160-P24 total immunoglobulin G (IgG) and its isotypes (IgG1, IgG2a and IgG2b) were monitored four weeks after the final injection using indirect enzyme-linked immunosorbent assay (ELISA). The mice immunized with the homologous DNA/B1 nanoparticles (G2 and G6) stimulated remarkably higher levels of total IgG, IgG1, IgG2a and IgG2b than with the naked DNA (G1 and G5; p < 0.001, Fig. 8a–d). In addition, the levels of IgG2a and IgG2b in the groups administrated by the homologous proteins (G3 and G7), and the heterologous DNA + B1 prime/protein + Montanide boost (G4 and G8) were enhanced as compared to other groups (p < 0.05; Fig. 8c, d). Our data indicated that all mice induced remarkably higher Nef-Vpu-Gp160-P24/Nef-Vif-Gp160-P24-specific total IgG, IgG1, IgG2a, and IgG2b as compared to control groups (G9-G12; p < 0.001, Fig. 8). The levels of IgG1 were lower in groups receiving B1 + nef-vif-gp160-p24 DNA (G6 and G8) as compared to B1 + nef-vpu-gp160-p24 DNA (G2 and G4), as well (p < 0.05).

Antibody responses against Nef-Vif-Gp160-p24 (a) and Nef-Vpu-Gp160-P24 (b) antigens: a Total IgG, b IgG1, c IgG2a and d IgG2b in different strategies: All analyses were performed in duplicate for each sample. The results were shown as mean absorbance at 450 nm ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
The IFN-γ and IL-5 production from splenocytes
Our data indicated that groups administrated by the DNA/B1 nanoparticles (G2 and G6) induced notably higher levels of IFN-γ and IL-5 as compared to groups receiving the naked DNAs (G1 and G5; p < 0.001; Fig. 9a, b). Surprisingly, the groups injected with the DNA/B1 nanoparticles (G2 and G6) could increase the levels of IL-5 higher than the rest of the groups (p < 0.05). However, the levels of IFN-γ were significantly higher in groups immunized with different regimens of Nef-Vif-Gp160-P24 (G5-G8) than groups immunized with the same regimens of Nef-Vpu-Gp160-P24 (G1-G4; p < 0.05, Fig. 9a). This result was reversed for IL-5 secretion (p < 0.05, Fig. 9b). In general, all groups secreted notably higher Nef-Vpu-Gp160-P24/Nef-Vif-Gp160-P24-specific IFN-γ and IL-5 cytokines than control groups (G9-G12; p < 0.001, Fig. 9a, b).

The levels of IFN-γ (a), IL-5 (b) and Granzyme B (c) in immunized groups with different formulations: The levels of IFN-γ (a), IL-5 (b) and Granzyme B (c) were measured using ELISA as mean absorbance at 450 nm ± SD for each set of samples. All analyses were performed in duplicate for each sample: ns: non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Granzyme B (GrB) secretion
Four weeks after the final injection (2nd booster), the Granzyme B secretion was determined. The data illustrated that all mice could enhance the secretion of Granzyme B significantly higher than the control groups (G9-G12; p < 0.001, Fig. 9c). Moreover, groups immunized by the DNA/B1 nanoparticles (G2 and G6) generated significantly higher levels of Granzyme B than groups immunized by the naked DNAs (G1 and G5; p < 0.001, Fig. 9c). The groups injected by heterologous immunization modalities (G4 and G8) could increase the secretion of Granzyme B significantly higher than the groups receiving homologous immunization modalities (G2 and G6; p < 0.001, Fig. 9c). Furthermore, the mice administrated by the Nef-Vif-Gp160-P24 multiepitope DNA and peptide constructs produced remarkably higher concentrations of Granzyme B than the mice injected by Nef-Vpu-Gp160-P24 multiepitope DNA and peptide constructs (p < 0.05, Fig. 9c).
Discussion
The non-viral delivery of DNA cargoes into mammalian cells is critical for therapeutic applications (Akinc et al. 2010). The delivery vehicles usually bind to negatively charged components of cell membranes, and then uptake into the cells (Wadia et al. 2004). Several delivery systems were offered to increase the induction of immune responses in vivo such as cationic polymers, virosomes, liposomes, nanoparticles, and cell-penetrating peptides (CPPs) (Jorritsma et al. 2016; Suschak et al. 2017). Cell-penetrating peptides (CPPs) penetrate into the cells to deliver biologically active cargoes. CPPs are short peptides (less than 30 amino acids) which are divided into cationic, amphipathic and hydrophobic categories based on their physicochemical properties (Kardani et al. 2019a, b). On the other hand, the engineered proteins involving cationic regions were developed to penetrate into the cells such as a penta-mutant GFP containing a patch of five arginine amino acids (Fuchs and Raines 2007). Moreover, the supercharged green fluorescent protein (scGFP) is a type of highly positively charged proteins which could deliver various cargoes in vitro (Motevalli et al. 2018; Shahbazi et al. 2018). In several studies, different variants of supercharged GFP were developed to transfer efficiently cargoes into mammalian cells (McNaughton et al. 2009). For instance, Lu et al. produced a series of supercharged GFP proteins ranging from − 30 to + 48 net theoretical charges which were able to deliver nucleic acids (Lawrence et al. 2007). Herein, B1 as one of the scGFPs was used to deliver DNA cargoes efficiently into mammalian cell lines. Previously, Simeon et al. demonstrated that B1 can be localized in the nucleus (Simeon and Chen 2013; Simeon et al. 2013).
In the current study, the levels of cellular and humoral immune responses were evaluated by various immunization strategies using two different multi-epitope constructs (Nef-Vpu-Gp160-P24 and Nef-Vif-Gp160-P24 antigens), and B1 cell-penetrating protein as a delivery system. At first, B1 protein was expressed efficiently in E.coli Rosetta expression strain, and purified under native conditions. In contrast, Simeon et al. could express the B1 protein in E.coli BL21 (DE3) strain (Simeon and Chen 2013; Simeon et al. 2013). However, protein-specific features such as temperature and time of induction can affect the yield of protein expression (Thompson et al. 2012). Moreover, our study indicated that B1 protein could form the stable nanoparticles with the nef-vpu-gp160-p24 and nef-vif-gp160-p24 multi-epitope DNA constructs at certain N/P ratio of 1:1. The B1 protein alone and in complex with multi-epitope DNA constructs was non-toxic at certain dose. We showed that B1 protein (+ 43 GFP) is stable in the presence of serum, and increases the serum half-life of plasmid DNA with which it is complexed. Indeed, a variant of + 43 GFP could mediate DNA transfection, enabling plasmid-based gene expression. This result was observed for DNA delivery using + 36 GFP (McNaughton et al. 2009). It was reported that superpositively charged proteins including + 36 GFP could penetrate into the mammalian cells with high potency in comparison to modestly cationic engineered proteins or cationic peptides (McNaughton et al. 2009). Recently, Bolhassani et al. confirmed that + 36 GFP can penetrate into cells efficiently in vitro and in vivo (Vahabpour et al. 2019). Moreover, Castanho et al. showed that the Dengue virus (DENV) capsid protein with a net charge of + 42 was able to deliver functional nucleic acids into mammalian cells (Freire et al. 2013). Our data demonstrated that B1 protein has notable efficiency to deliver both multi-epitope DNA constructs into HEK-293T cells. The flow cytometry results showed that the percentage of nef-vif-gp160-p24 and nef-vpu-gp160-p24 DNA expression was about 35.64% and 32.63%, respectively. These percentages were approximately 53.72% and 66.85% for TurboFect/pEGFP-nef-vpu-gp160-p24 and TurboFect/pEGFP-nef-vif-gp160-p24, respectively. In addition, the transfection efficiency of TurboFect/pEGFP-N1 and TurboFect/pEGFP-B1 was ~ 95.69% and ~ 68.55%, respectively. Indeed, eGFP replaced by B1 (pEGFP-B1) indicated that B1 can be expressed successfully in mammalian cells. On the other hand, B1 protein could penetrate into the cells about 60.19% without any transfection reagent. Thus, it is suitable to use B1 protein for delivery of other cargoes. The western blot analysis confirmed the potent transfection of both DNA constructs using B1 at N/P ratio of 1:1 as clear bands of about 32 kDa in vitro.
Different immunization strategies such as DNA/DNA, multi-epitope peptide/multi-epitope peptide, and DNA/multi-epitope peptide were used in BALB/c mice. Recently, it was reported that the heterologous prime-boost immunization strategy could enhance both humoral and cellular immunity in various laboratory animal models (Kardani et al. 2020, 2016; Stachyra et al. 2017; Valdés et al. 2019). Our data demonstrated that groups administrated by the DNA/B1 nanoparticles induced notably higher levels of IFN-γ, IL-5 and Granzyme B as compared to groups receiving the naked DNAs. Moreover, the levels of IFN-γ and Granzyme B were significantly higher in groups immunized with different regimens of Nef-Vif-Gp160-P24 than groups immunized with the same regimens of Nef-Vpu-Gp160-P24. On the other hand, the levels of IgG2a and IgG2b in groups administrated by the heterologous DNA + B1 prime/multi-epitope peptide + Montanide boost were enhanced similar to the homologous multi-epitope peptides + Montanide as compared to other groups. In our previous study, immunization of BALB/c mice using the heterologous DNA + HR9 or MPG CPP prime/multi-epitope peptide + CyLoP-1 or LDP-NLS boost modalities for both constructs could significantly increase the levels of IgG2a, IgG2b, IFN-γ, and Granzyme B directed toward Th1 immune responses as compared to homologous prime/boost with multi-epitope DNA or peptide constructs (Kardani et al. 2020). On the other hand, mice immunization with human papillomavirus (HPV) E7 protein/ + 36 GFP nanoparticles induced a higher Th1 cellular immune response with the predominant IgG2a and IFN-gamma levels than those induced by E7 protein, alone. It was observed that the E7 DNA/ + 36 GFP and E7 protein/ + 36 GFP nanoparticles similarly protected mice against TC-1 tumor challenge (~ 67%) as compared to E7 DNA and E7 protein (~ 33%) (Motevalli et al. 2018). Herein, we showed that B1 protein could induce Th1 cellular immune responses in heterologous DNA prime/multi-epitope peptide boost immunization. However, B1 protein could enhance the secretion of IgG1 and IL-5 in DNA immunization. Indeed, our data showed a mixture of Th1 and Th2 responses directed toward Th1 response especially in heterologous prime-boost regimens.
Conclusion
In summary, delivery of DNA cargoes into living cells using cell-penetrating proteins is an efficient approach for developing therapeutic vaccines via the formation of non-covalent complexes. Our data demonstrated that B1 protein complexed with both constructs drives T cell responses towards a Th1-type without side effects. These stable nanoparticles (DNA/B1 protein) with a lower dose of DNA plasmids (2 µg versus 50 µg of the naked DNA) increased the uptake of DNA constructs, and subsequently stimulated both humoral and cellular immune responses in immunized mice. However, further studies are required to determine its mechanism in vitro and in vivo as well as its efficiency in a challenge model.
References
Akinc A, Querbes W, De S et al (2010) Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther 18:1357–1364
Bolhassani A, Javanzad S, Saleh T, Hashemi M, Aghasadeghi MR, Sadat SM (2014) Polymeric nanoparticles: potent vectors for vaccine delivery targeting cancer and infectious diseases. Hum Vaccines Immunother 10:321–322
Bolhassani A, Kardani K, Vahabpour R et al (2015) Prime/boost immunization with HIV-1 MPER-V3 fusion construct enhances humoral and cellular immune responses. Immunol Lett 168:366–373
Brock DJ, Kustigian L, Jiang M et al (2018) Efficient cell delivery mediated by lipid-specific endosomal escape of supercharged branched peptides. Traffic 19:421–435
Cluck DB, Underwood RF (2018) A therapeutic perspective of living with human immunodeficiency virus/AIDS in 2017. Nursing Clinics 53:97–110
Crosio MA, Via MA, Cámara CI, Mangiarotti A, Del Pópolo MG, Wilke N (2019) Interaction of a polyarginine peptide with membranes of different mechanical properties. Biomolecules 9:625
Dings RP, Cannon M, Vang KB (2018) Design of gold nanoparticles in dendritic cell-based vaccines. Part Part Syst Charact 35:1800109
Freire JM, Veiga AS, Conceição TM et al (2013) Intracellular nucleic acid delivery by the supercharged dengue virus capsid protein. PLoS ONE 8:e81450
Freire JM, Almeida Dias S, Flores L, Veiga AS, Castanho MA (2015a) Mining viral proteins for antimicrobial and cell-penetrating drug delivery peptides. Bioinformatics 31:2252–2256
Freire JM, Santos NC, Veiga AS, Da Poian AT, Castanho MA (2015b) Rethinking the capsid proteins of enveloped viruses: multi-functionality from genome packaging to genome transfection. FEBS J 282:2267–2278
Fuchs SM, Raines RT (2007) Arginine grafting to endow cell permeability. ACS Chem Biol 2:167–170
Hasnain MS, Nayak AK (2019) Carbon nanotubes in vaccine delivery. Carbon nanotubes for targeted drug delivery. Springer, Singapore, pp 69–73
Jorritsma SH, Gowans EJ, Grubor-Bauk B, Wijesundara DK (2016) Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine 34:5488–5494
Kardani K, Bolhassani A, Shahbazi S (2016) Prime-boost vaccine strategy against viral infections: mechanisms and benefits. Vaccine 34:413–423
Kardani K, Milani A, Shabani SH, Bolhassani A (2019a) Cell penetrating peptides: the potent multi-cargo intracellular carriers. Expert Opin Drug Deliv 16:1227–1258
Kardani K, Hashemi A, Bolhassani A (2019b) Comparison of HIV-1 Vif and Vpu accessory proteins for delivery of polyepitope constructs harboring Nef, Gp160 and P24 using various cell penetrating peptides. PLoS ONE 14:e0223844
Kardani K, Hashemi A, Bolhassani A (2020) Comparative analysis of two HIV-1 multiepitope polypeptides for stimulation of immune responses in BALB/c mice. Mol Immunol 119:106–122
Kurzawa L, Pellerano M, Morris MC (2010) PEP and CADY-mediated delivery of fluorescent peptides and proteins into living cells. Biochim Biophys Acta 1798:2274–2285
Lawrence MS, Phillips KJ, Liu DR (2007) Supercharging proteins can impart unusual resilience. J Am Chem Soc 129:10110–10112
Li G, De Clercq E (2016) HIV genome-wide protein associations: a review of 30 years of research. Microbiol Mol Biol Rev 80:679–731
Ma YF, Yang YW (2010) Delivery of DNA-based cancer vaccine with polyethylenimine. Eur J Pharm Sci 40:75–83
McNaughton BR, Cronican JJ, Thompson DB, Liu DR (2009) Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc Natl Acad Sci USA 106:6111–6116
Motevalli F, Bolhassani A, Hesami S, Shahbazi S (2018) Supercharged green fluorescent protein delivers HPV16E7 DNA and protein into mammalian cells in vitro and in vivo. Immunol Lett 194:29–39
Rajapaksa AE, Ho JJ, Qi A et al (2014) Effective pulmonary delivery of an aerosolized plasmid DNA vaccine via surface acoustic wave nebulization. Respir Res 15:60
Sadeghian F, Hosseinkhani S, Alizadeh A, Hatefi A (2012) Design, engineering and preparation of a multi-domain fusion vector for gene delivery. Int J Pharm 427:393–399
Sardesai NY, Weiner DB (2011) Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol 23:421–429
Schwendener RA (2014) Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2:159–182
Shahbazi S, Haghighipour N, Soleymani S, Nadji SA, Bolhassani A (2018) Delivery of molecular cargoes in normal and cancer cell lines using non-viral delivery systems. Biotechnol Lett 40:923–931
Simeon RL, Chen Z (2013) A screen for genetic suppressor elements of hepatitis C virus identifies a supercharged protein inhibitor of viral replication. PLoS ONE 8:e84022
Simeon RL, Chamoun AM, McMillin T, Chen Z (2013) Discovery and characterization of a new cell-penetrating protein. ACS Chem Biol 8:2678–2687
Stachyra A, Pietrzak M, Macioła A et al (2017) A prime/boost vaccination with HA DNA and Pichia-produced HA protein elicits a strong humoral response in chickens against H5N1. Virus Res 232:41–47
Suschak JJ, Williams JA, Schmaljohn CS (2017) Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum Vaccines Immunother 13:2837–2848
Thompson DB, Cronican JJ, Liu DR (2012) Engineering and identifying supercharged proteins for macromolecule delivery into mammalian cells. Methods Enzymol 503:293–319
Vahabpour R, Basimi P, Roohvand F et al (2019) Anti-viral effects of superpositively charged mutant of green fluorescent protein. Protein Pept Lett 26:930–939
Valdés I, Izquierdo A, Cobas K et al (2019) A heterologous prime-boost strategy for immunization against Dengue virus combining the Tetra DIIIC subunit vaccine candidate with the TV005 live-attenuated tetravalent vaccine. J Gen Virol 100:975–984
Wadia JS, Stan RV, Dowdy SF (2004) Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft micropinocytosis. Nat Med 10:310–315
Acknowledgements
None
Author information
Authors and Affiliations
Contributions
Azam Bolhassani conceived and designed the experiments. Kimia Kardani, Azam Bolhassani and Atieh Hashemi reviewed all data, analyzed the results, and wrote the manuscript. Kimia Kardani and Elnaz Agi performed the experiments. Azam Bolhassani reviewed the manuscript and provided scientific advices.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Ethical approval
All mice were maintained under pathogen-free conditions, and the whole in vivo experiments procedures was approved by protocols and care of laboratory animals at Pasteur Institute of Iran (ethical code:1070).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kardani, K., Bolhassani, A., Agi, E. et al. B1 protein: a novel cell penetrating protein for in vitro and in vivo delivery of HIV-1 multi-epitope DNA constructs. Biotechnol Lett 42, 1847–1863 (2020). https://doi.org/10.1007/s10529-020-02918-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02918-w