
Abstract
In the paper the chemical modification of activated carbon was presented. The activated carbon was modified by nitric acid. For the tested activated carbons, the following physical and chemical properties were determined: bulk density, pH of the water extract, and effective diameter of the grains. Elemental and technical analyses were performed. The pore distribution was determined using mercury porosimetry. Low-temperature nitrogen isotherms were used to analyze the microporous structure. The chemical properties of the surfaces of the tested adsorbents were interpreted by means of the Boehm method. The water vapor adsorption isotherms at 303 K were determined and the adsorption capacity towards methanol was tested using the desiccator method. Thermogravimetric tests were used and, at the same time, the released oxidizing gases from the tested samples were analyzed using a mass spectrometer. As a result of the modification, activated carbon enriched with acidic oxygen functional groups was obtained. The resulting modified activated carbon showed worse structural parameters (when compred to umodified activated carbon), however it was characterized by a higher adsorption capacity in relation to polar adsorbates such as water and methanol, especially in the low pressure range.
Similar content being viewed by others
1 Introduction
Activated carbon is one of the earliest and most widely used adsorbents. Due to its peculiar properties such as: well-developed specific surface, considerable porosity, well-developed micro-, meso- and macroposity, and high capacity for selective sorption, activated carbons have been found as important materials used in various fields of industry.
The sorption-properties of carbonaceous adsorbents depends on the nature and structure of carbon. The physical and chemical properties of activated carbons may be modelled by means of various methods, i.e. the conditions of activation (temperature, the process time, the activating agents), the choice of the precursor and the application of additives [6].
The chemical properties of activated carbon depends mainly on the structure of the carbonaceous sorbent. The surface of activated carbon may be subjected to processes of modification in order to change and enhance its properties. It consists mainly in the introduction of functional groups on the surface of carbon materials or in their structure.
The methods of modification may be classified as chemical and physical. Chemical methods enable one to increase the amount of surface functional groups by means of the activity of oxidising or reductive agents. This modification may be conducted in the liquid or gas phase. Oxidising substances in the liquid phase usually include: inorganic acids (nitric acid, sulphuric acid, hydrochloric acid), hydrogen peroxide and organic acids [1, 7, 14]. Gas-based modification may be conducted by means of oxygen derived from air; water vapour; carbon dioxide; ozone and nitric oxides. Oxidising liquids are the most efficient, because it is easy to control the process and to achieve a high level of oxidation of the surface. In this way one obtains an increased amount only of acidic surface oxygen groups. One of the disadvantages of this method is associated with the fact, that the surface of the sorbent becomes polluted with reduced forms of an oxidant, which requires material purification, generating exceed amounts of wastes.
The acidic as well as the alkaline properties may be enhanced by gas oxidation, although this method is less efficient in comparison with oxidation by means of liquid substances.
The amount and the type of oxygen groups that are created depends mainly on the oxidant and the temperature at which modification is conducted. Modification in the liquid phase usually leads to the creation of large amounts of acidic oxygen groups [11, 16].
In the work [12] activated carbon was subjected to heat-related modification by means of the following substances: HNO3, H2O and HCl. The application of HNO3 allowed to the increase in the concentration of acidic oxygen groups on the surface of activated carbon, and it also increased the adsorption-related properties of the modified coal in relation to lead ions.
Modification of activated carbon may also be conducted by means of alkaline agents. Modification by means of potassium hydroxide considerably improves the physico-chemical properties of activated carbon [9]. One may obtain material that is characterised by a well-developed pore structure, considerable adsorption-related capacity in reference to methanol and material that is enriched with surface oxygen groups [5].
The modified material, enriched with oxygen groups, can be used to recover volatile organic solvents, to remove harmful substances from the liquid phase [10] and used in adsorptive cooling systems [15].
2 Experimental part
2.1 Materials
The work utilizes granulated activated carbon courtesy of the Global Carbon Solution company, produced by means of activation of the coconut shards by water vapour. Due to the highly-developed pore structure it is intended for adsorption in the gas phase. Commercial activated carbon (WA) was subjected to chemical modification. About 15 g of WA was mixed with 300 cm3 65% HNO3 diluted with steam water 1:1 in a round bottom flask with a heating mantle fitted with a reflux condenser. A mixture which was prepared in this way was kept at a boiling temperature for 3 h. Then the product was left to cool, filtered and rinsed with distilled water until a constant pH was obtained. The obtained activated modified carbon was denoted as WA1.
2.2 Characteristics of material
On the basis of the PN-85 C-97,555/10 norm, the pH of the water extract of the investigated materials was determined, using Elmetron CX-502 (ELMETRON, Zabrze, Poland). The bulk density and effective grains diameter of activated modified carbons were listed in Table 1.
In order to compare the granulation of the materials before and after modification, the analysis of the grain composition was studied. A sieve analysis was applied and the distribution of the grain sizes was shown in Fig. 1. On the basis of the data that was obtained, the effective diameter dz was calculated according to the following formula:
where: xi – mass fraction of the i-component.
di – grain diameter calculated as arithmetical diameter of the sizes of openings of both adjacent sieves.
The investigated materials were also characterized by means of elemental and technical analyses (Table 2). The content of the basic elements which are components of coal matter (C, H, S) was determined using ELTRA CHS 580 (Verder Scientyfic, Germany-Netherlands). The content of oxygen and nitrogen was calculated as by difference.
Humidity Wa (according to PN-80/G-04511), ash Aa (according to PN-80/G-04512), and volatile matter Va (according to PN/G-04516) contents as well as the heat of combustion Qs (according to PN/G-04513) were evaluated. The technical analysis was performed in the apparatus ELTRA TGA Thermostep (Verder Scientyfic, Germany-Netherlands).
The calorimeter (AC500 Isoperibol Calorimeter, LECO USA) was used for the determination of heat of combustion.
2.3 Structural properties
In order to study the changes of the texture, predominantly meso- and macropore texture, samples were subjected to mercury porosimetry analysis. This method involves the phenomenon of capillarity. Mercury, which (under normal pressure), after the prior degasification of the porous solid, does not enter the pores which radius exceed 7000 nm, is used as a pycnometric liquid.
In mercury porosimetry, the determination of the distribution of pores amounts to the establishment of the volume of mercury poured into the pores at a specific pressure. The relationship between the pressure p and the radius of the pores r filled with mercury is represented by Washburn’s equation.
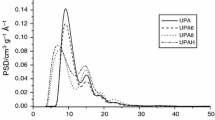
The changes in the pore structure, mainly of the macropores of activated carbons WA and WA1 was determined by means of a low-pressure PASCAL 140 and high-pressure PASCAL 440 (Milano, Italy) apparatuses. The equipment enables to determine the distribution of pores in a sample from a vacuum to a pressure of 150 MPa, which corresponds with the range of the radii of pores 5 ÷ 6⋅104 nm. The measurements were conducted upon grains. One obtained differential distributions of the volumes of pores according to their effective radii, which are presented in Fig. 2. The volumes of pores in the selected ranges of radii that were determined are listed in Table 3.
On the basis of porosimetric and densimetric research the porosity εT and the total volume of pores VT were determined (Table 4).
Low-temperature (77 K) isotherms of the adsorption/desorption of nitrogen were investigated, in order to perform an analysis of the micropore texture of the activated carbons. The measurements were conducted by means of Autosorb-1-C (Quantachrome Instruments, USA). Prior to the measurements, the samples were degasified at a temperature of 473 K for 12 h under vacuum conditions. On the basis of the data that was obtained, the volume of micropores (Vmic) and the average width of the pores (xDR) by applying the Dubinin-Radushkevich equation (DR) were calculated. The apparent specific surface area (SBET) was calculated by means of the Brunauer, Emmett and Teller equation. Data in reference to activated carbon WA were drawn from work [4]. The calculated parameters from porosimetric and adsorption-related research are listed in Table 4.
2.4 Chemical surface characterization
The chemical properties of the surfaces of the adsorbents under study were evaluated by means of the Boehm method, which is a general procedure applied for the purpose of determining surface functional groups [2, 8]. The Boehm titration method provides both qualitative as well as quantitative information about alkaline and acidic groups (in the carboxyl group, lactone and phenol). The acidity of these groups varies and they may be distinguished by neutralisation with various solutions: HCl (in reference to alkaline groups) as well as NaHCO3, Na2CO3 and NaOH (for acidic groups). The number of alkaline groups was determined by titration with a NaOH solution a surplus of nonreacted HCl. Reagents were used in the determinations (Avantor, Gliwice, Poland). The results that were obtained are presented in Table 5.
2.5 Adsorption capacity in reference to polar adsorbates
The presence of oxygen groups on the surface of carbon is clearly manifested in the case of the application of polar adsorbates. The most considerable relationships between sorption and the content of oxygen functional groups is observed in reference to sorbates, which may form hydrogen bonds. A water molecule is an example of such a sorbate, which enables to evaluate and determine the amount of surface oxygen groups. The adsorption of water vapour in porous carbon materials depends on the chemical structure of the surface and the shape and the size of pores.
In order to evaluate the polarity of the surfaces of the activated carbons under study, the isotherms of the sorption of water was determined by means of the liquid micro burette method. Samples of activated carbon were degassed in a vacuum 0.001 Pa to remove adsorbed molecules. Sorption isotherms of water vapors were measured using volumetric apparatus in a liquid microbourette apparatus at temperature of 303 K in the range of relative pressures p/p0 = 0.01–0.95. Apparatus with liquid microburretes allows to determine isotherms of sorption/desorption polar and apolar vapours at a pressure range from vacuum to the absolute vapour pressure. Before the sorption measurement the samples had to be properly prepared. Therefore, it is necessary to clean the surface of the samples from the previously absorbed gases and vapours, and also removing from the micropores other, adsorbed specific molecules. In addition, to get a better cleaning effect on the surface and porous structure of the research material, several rinsing with helium under pressure was performed. Helium atoms are not significantly being adsorb on the adsorbent surface and provide the necessary kinetic energy for the adsorbed particles vapours and gases, thereby knock them from the surface of the adsorbent. After each rinse helium, the sample was again degassed. The portion of adsorbate was added to the sample, and after reaching thermodynamic equilibrium (after 24 h) the amount of adsorbed gas was determined and then the next point of isotherm at higher pressure was measured. Measurements of the isotherms in a full range of relative pressures for vapor adsorbates. The results which were obtained are presented in Fig. 3.
In order to study the difference in the adsorption capacity in reference to polar adsorbates, the amount of adsorbed methyl alcohol vapours was determined. The impregnation of the samples of the activated carbons under study was conducted in statistical conditions by means of the so-called desiccation method. The measurement was carried out on samples in the amount of approx. 1 g ± 0.1 mg and inputed in desiccators. The samples were previously dried at 105 °C. The sample is being placed in a desiccator filled with methanol 99.9 wt% (Avantor, Gliwice, Poland) in amount 100 cm3. Next, the mass of the sample is being registered until a state of equilibrium corresponding to the saturation of the adsorbent is obtained. The temperature of the sample impregnation process was 296 K.
2.6 Thermogravimetric analysis
The methods of thermal analysis may be used in the research in the nature of the surface of activated carbons. During the heating of the sample one may observe physical and chemical changes. Measurements are usually conducted in a gaseous atmosphere under atmospheric pressure – in an oxidising atmosphere (O2, air) or a neutral one (N2, Ar). The (partial) loss of the mass of the sample that was studied may be caused by: desorption of absorbed gases (of water vapour, air); oxidation with the release of gas products; thermal breakdown with the release of gas products. The growth of the mass of the sample occurs when the substance under study reacts with a gas which flows through the furnace. A comparison of the changes of the mass of coal, heated at a particular rate with a simultaneous analysis of the gases which are being released enables one to evaluate the thermal stability of the particular functional groups.
Thermogravimetric analysis (TG) was conducted by means of a TGA Q5000 IR thermobalance (TA Instruments, USA). A change in the mass was registered in the temperature range of 25–900 °C with a rate of heating of 10 °C min− 1, in the flow of synthetic air (100 Nml·min− 1). On the basis of the results a differential curve (DTG) in a time function was determined.
At the same time, an analysis upon the oxidising gases which were released from the samples was determined by means of a ThermoStar GSD 301 T1 mass spectrometer (Pfeiffer Vacuum). The following gases production was recorded: H2O, NO, NO2, CO2, SO2 and SO3. The results which were obtained are presented in Fig. 5.
3 Results and discussion
As a result of HNO3 modification a carbonaceous material of acidic nature (pH 3.8) was obtained. A slight fragmentation of the modified carbon material was observed, which was reflected by the decrease in the value of the effective diameter of the grains and the increase in the bulk density (Table 1).
Activated carbon WA showed a relatively wide distribution of grains in the range of 1-5.5 mm. In reference to the WA1 adsorbent the fragmentation was lower, but still observable, which was reflected by the attenuation of the maximum at about 3.25 mm. The effective diameter which was calculated in reference to modified coal was lowered by 3% than for the initial activated carbon (WA). Chemical modification caused a slight fragmentation of the material, which influenced the increase in the value of bulk density by 13% in reference to carbon WA.

The distribution of the grain sizes of activated carbons
The elemental and technical analysis of activated carbons were done and the results are listed in Table 2. The total content of the atoms of oxygen and nitrogen in reference to modified carbon increased considerably, at the expense of the reduction of the content of elemental coal by about 30%. Commercially available activated carbon is characterised by a low amount of humidity (4.18%) and ash (1.77%).
By means of mercury porosimetry analysis obtained differential distributions of the volumes of pores according to their effective radii, which are presented in Fig. 2. Both carbons were characterised by a considerable number of pores of the ranges of pore radii 200 ÷ 2000 nm and a lack of macropores greater than 2000 nm. Modification by means of nitric acid did not affected the improvement of the pore structure of the carbon, even though one may clearly observe an increase in the content of pores in the range of 5 ÷ 30 nm (43% in the total content of pores). The nature of the course of the summary curve of the volume of pores (ascending curve) suggests that the samples feature pores of size r < 5 nm. As a rule the mercury porosimetry method enables to study the pores in the range of macro- and mesopores. The volumes of pores in the selected ranges of radii that were determined are listed in Table 3.

Histograms of the differential distribution of the volumes of pores according to the activated carbons effective radii
Commercially available activated carbon is characterised by very well-developed micro- and mesopore surface and a well-developed/high specific surface.
(1120 m2·g-1). As a result of modification by means of concentrated HNO3 acid a notacible changes of the structural parameters were observed. The apparent specific surface area SBET decreased by 37%. Also, both the volume of micropores and mesopores decreased, which was also confirmed in other publication [13]. The structural and densimetric parameters of activated carbons are presented in Table 4.
The initial activated carbon is an alkaline sorbent which features few lactone and phenol groups. The results obtained from the Boehm method confirmed the introduction of oxygen groups into the structure of the modified activated carbon WA1. All the types of the determined oxygen functional groups increased their share. The phenol and carboxyl groups were the substances whose greatest amounts were identified on the surface of modified WA1. The total amount of acidic groups for coal WA1 is 6.725 mmol·g-1 and for non-modified coal WA it was0.252 mmol·g-1. Along with the modification one can also observe a slight increase in the content of alkaline groups.
The shape of the isotherms of the adsorption of water vapour and its location enabled us to interpret the polarity of the surface and the pore structure of carbon. An experimental isotherm of water vapour for carbon WA manifests a typical course in reference to the isotherms of type-V adsorption according to IUPAC (Fig. 3). For low values of relative pressure, the adsorption was relatively low and it clearly increased from a relative pressure of about 0.5 p/p0. For modified carbon WA1, the water vapour adsorption curve was ascending in the whole range of relative pressures. For low relative pressures (up to the 0.6 p/p0) the amount of adsorbed water vapour was greater for modified carbon than it was for non-modified carbon. As reported in the literature [3], in the initial range of relative pressures the adsorption of polar water molecules happens mostly in the original centres of sorption, which constitute surface oxygen functional groups. Surface oxygen functional groups are polar centres of sorption, which means that the sorption of water vapour on activated carbons is polar in nature.

Water vapour adsorption isotherms in 303 K
Using the static-desiccator method, methanol sorption capacity were empirically determined on the test material at room temperature 296 K. The maximum amount of adsorbed methanol, in the amount of 31.88 wt%, for the WA1 carbon was obtained after the first day. For unmodified WA carbon, the maximum amount was reached after 4 days and was 28.08 wt%. In reference to the carbons that were studied the equilibrium is established after 4 days.
Referring to modified carbon, it can be observed, that the adsorption capacity of methanol was greater and the state of equilibrium in reference to this carbon was obtained in a shorter period of time, which confirms the high adsorption capacity for polar sorbents.
The extremum on the DTG curve and the inflection point on the TG curve corresponds with the maximum rate of the change of mass. A slight extremum may be observed around the 600 °C for both WA and WA1 activated carbons. By comparing thermogravimetric curves (Fig. 4; Table 6) it can be observed, that the (partial) loss of mass in reference to the modified carbon WA1 was greater in the range 25–180 °C and that it undergoes almost complete combustion at a temperature of up to 600 °C. In reference to non-modified carbon WA, initially the (partial) loss of mass is slighter, and the greater part of the mass is diminished in the range of 180–600 °C.
TG and DTG curves are presented in Fig. 4, and the calculated losses of mass in the selected temperature ranges are presented in Table 6.

The mass loss curve (TG) and a differential curve (DTG) in temperature function
By analysing the spectra of the mass spectrometer (Fig. 5) it can be seen, that in reference to modified carbon the intensity of H2O in the temporal range 60–90 min was much greater, which corresponds with the temperature range 40–320 °C. The release of SO2 and SO3 oxides up to 110 min was also greater. This attests to the strong oxidising properties of the modifier that was applied (HNO3). In reference to non-modified carbon, a greater amount of CO2 was registered as the WA featured a much greater amount of elemental coal, as well as nitric oxides. Modification conducted with a strong oxidising compound (HNO3) does not cause the release of nitrogen atoms but it does oxidise the surface, enriching it with groups which contain oxygen.

The content of the oxides which were released
4 Conclusions
The chemical method of modification that is proposed herein, changes both the structural and the surface properties of activated carbon. Modified carbon WA1 underwent slight fragmentation, which caused an increase in bulk density (0,5433 g·cm− 3) and a decrease in total porosity (0.295 cm3·cm− 3). By surface modification of the activated carbon with a strong acid, an increase in the number of pores in the range of 5–30 nm occured, although the general structural parameters, such as specific surface and the volume of micro- and mesopores were reduced.
As a result of the modification the nature of carbon changed from alkaline to acidic. Commercially available activated carbon features a small amount of lactone and phenol groups – a total of 0.252 mmol·g− 1. As a result of modification the WA1 carbon contains a considerable amount of carboxyl, phenol and lactone groups. The total amount of oxygen functional groups which are acidic was 6.275 mmol·g− 1. The amount of groups which are alkaline was slightly increased (by 4%).
In this research, a considerable amount of oxygen functional groups, which altered the chemical nature of the adsorbent was reported.
Modified carbon was characterised by a greater adsorption capacity in reference to polar adsorbates such as water and methanol, especially in the range of low pressures.
The authors would like to thank the AGH University of Science and Technology, Krakow, Poland for financial support (No. 16.16.210.476).
Data Availability
Not applicable.
References
Bagreev, A., Locke, D.C., Bandosz, T.J.: H2S adsorption/oxidation on adsorbents obtained from pyrolysis of sewage-sludge-derived fertilizer using zinc chloride activation. Ind. Eng. Chem. Res. 40, 3502–3510 (2001). https://doi.org/10.1021/IE010165W/ASSET/IMAGES/LARGE/IE010165WF00011.JPEG
Boehm, H.P.: Surface oxides on carbon and their analysis: A critical assessment. Carbon N. Y. 40, 145–149 (2002). https://doi.org/10.1016/S0008-6223(01)00165-8
Brennan, J.K., Bandosz, T.J., Thomson, K.T., Gubbins, K.E.: Water in porous carbons. Colloids Surf. Physicochem Eng Asp. 187–188, 539–568 (2001). https://doi.org/10.1016/S0927-7757(01)00644-6
Brodawka, E., Bałys, M., Szczurowski, J., Czepirski, L., Zarębska, K., Da Costa, P.: Nanoporous carbonaceous adsorbents for enrichment of ventilation air methane (VAM) with methane. Presented at the (2019)
Buczek, B., Wolak, E.: Potassium hydroxide modified active carbon for adsorptive refrigerators. Adsorption. 14, 283–287 (2008). https://doi.org/10.1007/S10450-007-9094-5/METRICS
Chand, R., Meenakshi, B., Roop, G., Bansal, C., Goyal, M.: Activated Carbon Adsorption. (2005). https://doi.org/10.1201/9781420028812
El-Hendawy, A.N.A.: Influence of HNO3 oxidation on the structure and adsorptive properties of corncob-based activated carbon. Carbon N. Y. 41, 713–722 (2003). https://doi.org/10.1016/S0008-6223(03)00029-0
Kalijadis, A.M., Vukčević, M.M., Jovanović, Z.M., Laušević, Z.V., Laušević, M.D.: Characterisation of surface oxygen groups on different carbon materials by the Boehm method and temperature-programmed desorption. J. Serbian Chem. Soc. 76, 757–768 (2011). https://doi.org/10.2298/JSC091224056K
Mokrzycki, J., Magdziarz, A., Rutkowski, P.: The influence of the Miscanthus giganteus pyrolysis temperature on the application of obtained biochars as solid biofuels and precursors of high surface area activated carbons. Biomass Bioenerg. 164, 106550 (2022). https://doi.org/10.1016/J.BIOMBIOE.2022.106550
Pamidimukkala, P.S., Soni, H.: Efficient removal of organic pollutants with activated carbon derived from palm shell: Spectroscopic characterisation and experimental optimisation. J. Environ. Chem. Eng. 6, 3135–3149 (2018). https://doi.org/10.1016/J.JECE.2018.04.013
Pietrzak, R., Wachowska, H., Nowicki, P.: Preparation of nitrogen-enriched activated carbons from brown coal. Energy and Fuels. 20, 1275–1280 (2006). https://doi.org/10.1021/EF0504164/ASSET/IMAGES/MEDIUM/EF0504164E00001.GIF.
Repelewicz, M., Choma, J.: Study of physicochemical properties of chemically modified activated carbons. Eng. Prot. Environ. 8, 5–17 (2005)
Repelewicz, M., Jedynak, K., Choma, J.: Porous structure and Surface Chemistry of active carbons modified with inorganic acids. Ochr. Środowiska. 31, 45–50 (2009)
Tang, L., Li, L., Chen, R., Wang, C., Ma, W., Ma, X.: Adsorption of acetone and isopropanol on organic acid modified activated carbons. J. Environ. Chem. Eng. 4, 2045–2051 (2016). https://doi.org/10.1016/J.JECE.2016.03.031
Wolak, E., Vogt, E., Szczurowski, J.: Modification of activated carbons for application in adsorption cooling systems. Tech. Sci. / Univ. Warm Maz Olsztyn nr. 22(1), 87–98 (2019)
Wu, C., Li, L., Zhou, H., Ai, J., Zhang, H., Tao, J., Wang, D., Zhang, W.: Effects of chemical modification on physicochemical properties and adsorption behavior of sludge-based activated carbon. J. Environ. Sci. 100, 340–352 (2021). https://doi.org/10.1016/J.JES.2020.08.005
Funding
Work carried out under Research subsidy of AGH, No. 16.16.210.476.
Author information
Authors and Affiliations
Contributions
Most of the experimental studies were carried out by two authors. Eliza Wolak used thermogravimetric analysis and wrote the main manuscript text. Agnieszka Orzechowska-Zięba used porosimetric method and edit the text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
I declare that the authors have no competing interests as defined by Springer, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wolak, E., Orzechowska-Zięba, A. Change of the surface and structure of activated carbon as a result of HNO3 modification. Adsorption 30, 121–128 (2024). https://doi.org/10.1007/s10450-023-00401-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-023-00401-2