
Abstract
Nineteen red deer areas in a densely populated region with a huge network of fenced motorways and the division into administrative management units (AMUs) with restricted ecological connectivity were investigated. In the season 2018/2019, a total of 1291 red deer samples (on average 68 per area) were collected and genotyped using 16 microsatellite markers. The results show a clear genetic differentiation between most of the AMUs. Fourteen AMUs may be combined into four regions with a considerable internal genetic exchange. Five areas were largely isolated or showed only a limited gene flow with neighbouring areas. Ten of the 19 AMUs had an effective population size below 100. Effective population sizes greater than 500–1000, required to maintain the evolutionary potential and a long-term adaptation potential, were not achieved by any of the studied AMUs, even when AMUs with an appreciable genetic exchange were aggregated. Substantial genetic differentiation between areas can be associated with the presence of landscape barriers hindering gene flow, but also with the maintenance of ‘red deer–free’ areas. Efforts to sustainably preserve the genetic diversity of the entire region should therefore focus on measures ensuring genetic connectivity. Opportunities for this goal arise from the establishment of game bridges over motorways and from the protection of young male stags migrating through the statutory ‘red deer–free’ areas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Red deer (Cervus elaphus) populations in continuous landscapes are in active genetic exchange with each other as long as the younger stags are able to migrate. In Germany, the law is indirectly limiting migration (German hunting law as amended in the year 2015 and the Hessian hunting law as amended in 2016). Twenty Hessian red deer areas were created in the 1950s as administrative red deer management units (AMUs) to address the high levels of damage to vegetation at that time. Shooting plans were issued for these AMUs, based in particular on the quantity of bark-stripping in beech and spruce. The AMUs are located in forested areas where red deer populations traditionally occurred. They represent essentially the remains of historical summer habitats of the red deer, while the former winter habitats in the floodplains are dissolved or no longer accessible. In the ‘red deer–free’ areas between AMUs, establishment of red deer populations should be prevented to protect vegetation from damage. Hunting in these areas could impede the migration of red deer between AMUs (Herzog et al. 2020). Moreover, the high human population density in Central Europe and the fragmentation of the landscape with urban sprawl and highways create obstacles that can severely inhibit genetic exchange between subpopulations (Hartl et al. 2003; Frantz et al. 2012).
Isolated populations suffer from genetic drift (Slatkin 1987). Especially in small isolated populations (Whitlock 2000), rare gene variants can be lost, often already with the loss of individual animals (Sperlich 1988; Balloux and Lugon-Moulin 2002). Allele losses and genetic impoverishment reduce the degree of heterozygosity (Frankham 2008; Stopher et al. 2012; Mukesh et al. 2013) and can lead to inbreeding depression (Slate et al. 2002; Walling et al. 2011). Recently, we have described the decline in genetic diversity of a small isolated AMU in Germany from the 1980s to the 2010s (Willems et al. 2016). This result and one of the highest densities of traffic and settlement areas for Germany led to the hypothesis of an interrupted gene flow between the Hessian AMUs. This hypothesis should be tested based on methods uncovering the genetic population structure and differentiation of the 19 main Hessian AMUs with the present study. If the hypothesis was confirmed, this should provide information to enable the responsible authorities to take appropriate countermeasures.
Methods
Red deer population and sampling
The study area covers the entire region of the federal state of Hesse in the centre of Germany (Fig. 1) with a north-south extension of 260 km; a west-east extension of 170 km; and a total area of about 21,000 km2. Human population density in 2018 was approximately 297 people per km2 (Statistisches Bundesamt) compared to the German average with 237 people/km2. The state comprises a mosaic of different types of land use, predominantly forest (42.5%), pastures, and agriculture. Hesse is the federal sate in Germany with the largest area of forest. Twenty AMUs are scattered across the state but vary substantially in size from 105 to 940 km2 (Table 1). Nineteen of the AMUs were studied. An extremely small AMU in the north, the Upland, an appendage to the larger AMUs of the neighbouring country North Rhine-Westphalia, was no longer hunted and therefore not sampled. Distances between AMUs range from 12.5 to 240 km (centre to centre). They are separated by settlement areas, fenced motorways, country roads, and eventually by larger contiguous farmland which may form barriers to the migration of red deer (Perez-Espona et al. 2008; Frantz et al. 2012). According to the local authorities and the red deer conservation societies, who are legally entrusted with the management of the respective AMUs, the estimated population size of the red deer management units in spring ranges from 70 animals in Wattenberg-Weidelsburg to 2400 animals in the Spessart (Table 1). However, these numbers are based on estimates, while exact numbers are not available. The estimates are based on recorded browsing damage, snow counts, and local hunters’ knowledge of the animals in their hunting grounds.

Red deer administrative management units (AMU) (yellow areas; black letters) in Hesse: Burgwald-Kellerwald (BKW), Dill-Bergland (DB), Gieseler Forst (GF), Hinterlandswald (HW), Hoher Vogelsberg (HV), Knuell (KNU), Krofdorfer Forst (KF), Lahn-Bergland (LB), Meissner-Kaufunger Wald (MKW), Noerdlicher Vogelsberg (NV), Odenwald (OD), Platte (PL), Reinhardswald (RW), Riedforst (RF), Rothaargebirge (RG), Seulingswald (SW), Spessart (SP), Taunus (TA), *Upland (UL, not investigated), Wattenberg-Weidelsburg (WW). Towns (red areas and red letters): Darmstadt (DA), Frankfurt (F), Fulda (FD), Giessen (GI), Hanau (HU), Kassel (KS), Limburg (LM), Marburg (MR), Offenbach (OF), Wetzlar (WZ), Wiesbaden (WI). Motorways: red lines, blue labelling
Samples were taken from tissues of red deer after hunting by the local district foresters. They were labelled, provided with information about the animal (sex and age class) and the hunting site, and frozen until they were processed in the laboratory. A total of 1291 samples were taken during the hunting season 2018/2019. We used samples from legally harvested animals that were provided by the hunters. No animals were killed specifically for the study. No living animals were sampled and no dropping antlers were sought or collected for the study. Because of the effects of sample size on the accuracy of the population genetic results (Reiner et al. 2019), the target was set to collect 60 samples per area. In the end, an average of 67.9 samples per area was collected. One area (Reinhardswald) was sampled particularly intensively with 204 samples that were analysed for a different purpose in a parallel study (Reiner et al. 2020). The minimum sample size was 47 samples in Hoher Vogelsberg (for details see Table 1).
DNA extraction and genotyping
DNA was extracted by using a commercially available kit (Instant Virus RNA Kit, Analytik Jena, Germany). For this purpose, 30 to 50 mg of tissue was processed according to the manufacturer’s instructions. DNA concentration was determined photometrically and adjusted to 5 ng/μl with RNAse-free water. The presence of high molecular weight DNA was confirmed by agarose gel electrophoresis. Sixteen microsatellites were used to genotype red deer as described in detail by Willems et al. (2016). Primers were combined in four multiplex PCRs (Supplementary Table 1). PCR was performed in a volume of 10 μl consisting of 5 μl of 2× Multiplex Mastermix (Qiagen, Germany), 4 μl of primermix, and 1 μl (5 ng) of extracted DNA. DNA was amplified after an initial denaturing step of 15 min in 26 cycles of denaturing at 94 °C for 30 s, annealing at 56 °C (multiplex PCR 4 at 50 °C) for 90 s, and extension at 72 °C for 30 s. After a final step at 60 °C for 30 min, PCR reactions were cooled down to 4 °C.
Capillary electrophoresis
One microliter of the fluorescently labelled PCR product and 0.375 μl DNA Size Standard 500 Orange (Nimagen, Netherlands) were added to 12 μl Hi-Di-formamide (Thermo Fisher Scientific, Germany) and electrophoresed on an ABI PRISM 310 automatic sequencer. Allele sizes were determined with the Peakscanner 2.0 software (Thermo Fisher Scientific, Germany).
Analysis of population genetic parameters
Most of the population genetic analyses were performed within the statistical software R (R Core Team 2017). Frequencies of null alleles were calculated with the function null.all implemented in the R package PopGenReport v3.0.4 (Adamack and Gruber 2014). Because the frequency of missing data was below 5%, null allele frequencies were estimated with the method described by Brookfield (1996). The 95% confidence interval (CI) was computed with 1000 bootstraps. If the 95% CI includes zero, null allele frequencies do not significantly differ from zero.
Deviations from the Hardy-Weinberg equilibrium (HWE) were tested with the function hw.test implemented in the R package pegas v0.12 (Paradis 2010). The test was performed as an exact test based on Monte Carlo permutations (n = 1000) of alleles (Guo and Thompson 1992).
Private alleles and evenness of allele distribution were determined with functions implemented in the R package poppr v2.8.3 (Kamvar et al. 2014).
Population genetic parameters (mean number of alleles, rarefied allelic richness, observed heterozygosity, expected heterozygosity, inbreeding coefficient Fis) were calculated with the function divBasic implemented in the R package diveRsity v.1.9.90 (Keenan et al. 2013). Fis values were given with their 95% CI obtained after 1000 bootstrap iterations.
The same R package was used to determine pairwise population differentiation using Fst (Weir and Cockerham 1984) and Jost’s D (Jost 2008) as metrics. Significance of differences in pairwise comparisons was assessed by 1000 bootstrapping iterations. Whereas Fst reflects demographic processes and fixation, Jost’s D is a measure of allelic differentiation (Jost et al. 2018).
The effective population size (Ne) was estimated with NeEstimator V2.1 (Do et al. 2014). Estimates of Ne were calculated with the linkage disequilibrium method with random mating as mating system. To exclude single-copy alleles, the critical value for the allele frequency (Pcrit) was set to 0.02 for populations with less than 50 and to 0.01 for 50 and more sampled individuals. To estimate the effect of different allele frequency thresholds, we calculated Ne for Pcrit values of 0.05, 0.02, and 0.01, and by omitting all allele singletons. Additionally, for comparison, Ne was determined from demographic data provided by the local authorities and by the local red deer conservation societies. Assuming a constant sex ratio of reproducing animals and no fluctuations in population size, Ne was calculated according to Wang et al. (2016) under the assumption of harem polygamy as mode of reproduction from the number of reproducing males (Nm) and females (Nf) as Ne = (4 * Nm * Nf)/(2 * Nm + Nf). To account for possible error rates in the estimates, Ne values were recalculated for Nm/Nf ratios varying between 0.7 and 1.3 times the estimated Nm/Nf ratio. The percentage of annual increase in inbreeding (dF) was calculated as 1/(2 * Ne).
To evaluate the genetic structure of the population, we used STRUCTURE 2.3.4 (Pritchard et al. 2000) and DAPC (Jombart et al. 2010). STRUCTURE uses a Bayesian model-based clustering method with a heuristic approach for estimating the number of clusters (K). STRUCTURE analysis was performed with K = 1 to 15 clusters assuming admixture and correlated allele frequencies. For each K, 10 independent runs with 100,000 burn-in and 200,000 MCMC iterations were performed. The optimal number of K was determined by the method of Evanno et al. (2005) using the software STRUCTURE harvester (Earl and von Holdt 2012). Likelihoods of cluster memberships were averaged over the ten runs with the online programme CLUMPAK (Kopelman et al. 2015). The most likely K-value determined by STRUCTURE harvester was only recognised as a crude measure to describe the level of genetic structure patterns. To identify underlying nested clusters, hierarchical STRUCTURE analysis was performed (Pritchard and Wen 2003; Edelhoff et al. 2020) by using the clusters of previous runs as input and setting the ‘LOCPRIOR’ as sampling location. Subsequent analyses were done only on the clusters, identified in the previous run. This approach was repeated until there was no further differentiation.
Population structure was additionally evaluated with the clustering procedure used in Discriminant Analysis of Principal Components (DAPC), implemented in the R package adegenet v2.0.1 (Jombart 2008; for a tutorial see http://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf). In a first step, a partitioning analysis was performed with K = 2 to 20 to detect the optimal number of clusters. Assuming an island model, the improvement of fit was monitored through the BIC (Bayesian information criterion) value which decreases until it reaches the optimal K. Results from the K-means procedure were used as input data for DAPC. The number of retained principal components was validated with the cross-validation function xvalDapc. Frequencies of individuals belonging to the different clusters were determined for each AMU and presented as pie charts. For clarity, DAPC clusters were shown in three maps emphasising clusters with regional distribution, spatially limited occurrence, and supra-regional spread.
To give a comprehensive overview of the genetic similarity between neighbouring AMUs, results from DAPC were transformed into percentages expressing the probability that individuals from two AMUs belonged to the same DAPC cluster (see Supplementary Table 2 for details). Genetic similarities were standardised by setting the two AMUs with the highest genetic similarity to 100%. This metrics was called relative genetic similarity and was calculated for all pairs of AMUs.
Results
Null allele frequencies of markers significantly different from zero were detected for nearly all AMUs except DB, HV, HW, and SP. The most prominent markers prone to null alleles were RT6 and BM4208 with null allele frequencies ranging from 7.9 to 21.2% (12.7 ± 4.3) and 7.1 to 12.0% (10.0 ± 1.6), respectively. All other null allele frequencies significantly different from zero were distributed across different markers and different AMUs and ranged from 4.7 to 14.2% (8.7 ± 2.7). We compared Fst calculations with and without considering null alleles using the software FreeNA (Chapuis and Estoup 2007). There were no significant differences between Fst values with and without including null alleles. Therefore, all loci were maintained.
Marker NVHRT48 had the highest (n = 31) and markers CSSM22N and CSSM14 the lowest number of alleles (n = 3).
A total of 95 private alleles were found, most of which were detected for markers NVHRT48 (n = 29), RT1 (n = 25), and T501 (n = 19). Private alleles were predominantly spread over three AMUs (RW, n = 35; OD, n = 30; SP, n = 14). No private alleles were found in BKW, KF, KNU, LB, MKW, PL, RF, SW, and WW.
Alleles of marker CSSM16 (n = 6) were most evenly distributed (evenness = 0.88) over all AMUs whereas allele frequencies of marker RT1 with 14 alleles varied substantially (evenness = 0.47). Although observed heterozygosity (Ho) of markers was consistently lower than expected heterozygosity (He), differences were statistically not significant (p = 0.7). None of the markers showed a consistent deviation from HWE.
Population genetic parameters of the AMUs are given in Table 2. The mean number of alleles (7.25 ± 0.73) varied between 5.7 (PL) and 8.9 (RW). Allelic richness (Ar) was highest for the SP (Ar = 7.5) and lowest for the PL (Ar = 5.3) AMU. Observed heterozygosity (Ho) varied between 0.61 and 0.7. Fis values ranged from − 0.007 (HW) to 0.076 (GF). Fis values significantly different from zero were detected for AMUs GF, HT, KNU, RG, RW, and SP. Inbreeding coefficients for GF, HT, and RW were significantly higher than those for HW and SW.
Six of the AMUs (GF, HV, KF, NV, PL, and WW) had a 95% CI including an estimated Ne calculated from genetic data smaller than 50 and thus an annual increase in inbreeding of greater than 1% (Table 3). All Ne values except for RW were identical for allele frequency thresholds as given in the ‘Methods’ section and calculated with ‘no allele singletons tolerated’ (Table 3). Therefore, all further statements are based on Ne calculated with the latter option. The annual increase in inbreeding was most pronounced for WW, NV, KF, and GF with a mean dF of 1.61%, 1.35%, 1.14%, and 1.10%, respectively. Effective population size and annual increase in inbreeding calculated with the Ne estimator correspond well with those estimated from demographic data (Table 4). Even with an error rate of 10 to 20% of the estimated Nm/Nf ratio, most of the demographic Ne of the AMUs lie within the 95% CI of the genetic Ne. Only demographic Ne for HV, HW, PL, SP, and WW were not included in the 95% CI of the genetic Ne. SP was found to have the highest Ne and thus the lowest annual increase in inbreeding. An annual increase in inbreeding of more than 1% was found for WW (1.91%), NV (1.30%), PL (1.28%), HV (1.26%), and KF (1.14%). None of the AMUs had an optimal Nm/Nf ratio of 0.5 at which Ne is maximal. Ratios ranged from 0.02 (HW) to 0.29 (BKW).
Hierarchical STRUCTURE analysis classified the 1291 individuals into four clusters on a first level (Fig. 2; Supplementary Figures 1 and 2). The Dirichlet parameter (α = 0.0531) indicated that there was limited admixture. On a second level, the four previous clusters were split into 2, 2, 2, and 4 further clusters, respectively (Supplementary Figures 3–10). The informativeness r of the LOCPRIOR was always below 1, meaning that the information of the sampling location was useful for the assignment of individuals to clusters.

Clustering of the red deer administrative management units (AMUs) in two consecutive steps (level 1 and 2) by hierarchical STRUCTURE analysis
In DAPC clustering (K = 2–20), minimum BIC values were observed with K = 15. Therefore, DAPC analysis was performed with 15 clusters and 80 retained principal components which explain 91.6% of the genetic variance. DAPC clusters corresponded very well with those determined with STRUCTURE. However, the additional five clusters revealed even more details of the underlying population structure. Figure 3 shows the distribution of gene clusters dividing the Hessian AMUs into four regions. Majority of individuals of the southeastern part of the state comprising the neighbouring AMUs Taunus, Platte, and Hinterlandswald were assigned to cluster 1. Hoher Vogelsberg (cluster 11), Noerdlicher Vogelsberg (clusters 4 and 11), Spessart (clusters 4 and 11), Seulingswald (cluster 4), and Gieseler Forst (clusters 4 and 11) in the southeast comprised individuals with similar genetic properties. Cluster 7 (Rothaargebirge, Dill-Bergland, and Lahn-Bergland) and 13 (Rothaargebirge, Dill-Bergland, Lahn-Bergland, Burgwald-Kellerwald, and Wattenberg-Weidelsburg) were predominant in the northwestern AMUs. A minor part of the animals of the neighbouring Krofdorfer Forst was also found in cluster 7. Cluster 12 only occurred in the north-east, with the closely neighbouring areas Riedforst and Meissner-Kaufunger-Wald.

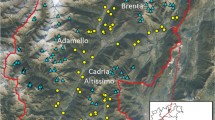
Major genetic clusters with regional distribution linking red deer administrative management units (AMUs) (clusters 1, 4, 7, 11, 12, and 13). The circle segments show the proportion of individuals of the particular AMU assigned to the respective cluster. White circle segments show the proportion of individuals belonging to different clusters (see Figs. 4 and 5). The exact percentages can be found in Table 5. Irregular yellow areas indicate the location and spread of the AMUs. Red areas represent larger urban areas, red lines represent motorways. BKW, Burgwald-Kellerwald; DB, Dill-Bergland; GF, Gieseler Forst; HV, Hoher Vogelsberg; HW, Hinterlandswald; KF, Krofdorfer Forst; KNU, Knuell; LB, Lahn-Bergland; MKW, Meissner-Kaufunger-Wald; NV, Noerdlicher Vogelsberg; OD, Odenwald; PL, Platte; RF, Riedforst; RG, Rothaargebirge; RW, Reinhardswald; SP, Spessart; SW, Seulingswald; TA, Taunus; WW, Wattenberg-Weidelsburg
The geographically limited occurrence of clusters is shown in Fig. 4. Gene clusters 2, 6, 8–10, 14, and 15 essentially only occurred in one or two AMUs. Cluster 2 contained predominantly individuals of the Knuell whereas animals of the Odenwald were almost exclusively classified into cluster 9. Cluster 6 was shared by individuals of the Reinhardswald and Knuell, and cluster 15 by animals of the AMUs Wattenberg-Weidelsburg and Krofdorfer Forst. Clusters 8 and 14 were preferentially found in the Reinhardswald, and cluster 10 in the Lahn-Bergland.

Geographically limited genetic clusters (comprising mostly one or two red deer administrative management units (AMUs) with a significant proportion of animals in the respective AMU. The circle segments show the proportion of individuals in the particular AMU assigned to the respective cluster. White circle segments show the proportion of individuals belonging to the clusters in Figs. 3 and 5
Clusters 3 and 5 show a particularly striking supra-regional distribution (Fig. 5). They were present in all AMUs either solitary or in combination. Cluster 3 was predominant in the Seulingswald as was cluster 5 in the Spessart.

Supra-regional spread of genetic clusters 3 and 5 with the participation of fewer individuals per red deer administrative management unit (AMU). The circle segments show the proportion of individuals in the particular AMU assigned to the respective cluster. White circle segments show the proportion of individuals belonging to the clusters in Figs. 3 and 4
As an easy and comprehensible way of comparing AMUs, the relative genetic similarity between neighbouring areas, based on the probability that individuals from two regions belong to the same DAPC clusters, is presented in Fig. 6. The maximum similarity was achieved when comparing Hinterlandswald and Platte (set to 100%). A comparably high level of agreement was found for the neighbouring areas Hoher Vogelsberg/Noerdlicher Vogelsberg, Hoher Vogelsberg/Gieseler Forst, and Meissner-Kaufunger-Wald/Riedforst (dark and light blue circles with similarities above 60%). The most massive barriers for red deer in the state are shown by the red circles. Here the degree of agreement between neighbouring areas was up to 15% of the maximum value. They run like red lines from north-east to south-west and from south-east to north-west, separating regions with several AMUs. Odenwald, Reinhardswald, and Knuell were clearly isolated. Odenwald showed by far the highest degree of isolation. Even between the areas with higher degrees of similarity, clear differences were still evident as shown by the orange (similarity between 15 and 30%) and light green circles (similarity between 30 and 45%). These results agree well with the results of the hierarchical structure analysis, but at the same time help to quantify the qualitative classification of the areas. Table 5 shows the corresponding relative genetic similarity between all Hessian AMUs. The correlation between Fst and relative genetic similarity (Perason’s r) was 0.691.

Relative genetic similarity of neighbouring red deer administrative management units (AMUs) (data are given as percentage of the maximum achieved similarity set to 100% between Hinterlandswald and Taunus). Relative genetic similarity is presented in six groups with 0 to 15% (red circles), > 15 to 30% (orange circles), > 30 to 45% (light green circles), > 45 to 60% (dark green circles), > 60 to 75% (light blue circles), and more than 75% (dark blue circles). Irregular yellow areas indicate the location and spread of the AMUs. Red areas represent larger urban areas, red lines represent motorways. BKW, Burgwald-Kellerwald; DB, Dill-Bergland; GF, Gieseler Forst; HV, Hoher Vogelsberg; HW, Hinterlandswald; KF, Krofdorfer Forst; KNU, Knuell; LB, Lahn-Bergland; MKW, Meissner-Kaufunger-Wald; NV, Noerdlicher Vogelsberg; OD, Odenwald; PL, Platte; RF, Riedforst; RG, Rothaargebirge; RW, Reinhardswald; SP, Spessart; SW, Seulingswald; TA, Taunus; WW, Wattenberg-Weidelsburg
There was also good agreement between pairwise Fst and Jost’s D values (r = 0.930; Table 6). Fst and Jost’s D values were normally distributed with a mean of 0.067 ± 0.020 (standard error) and 0.130 ± 0.040, respectively. The pairwise comparison BKW/RG had the lowest values (Fst: 0.015; Jost’s D: 0.027). Maximum values were found for the pairwise comparisons OD/KF (Fst: 0.122; Jost’s D: 0.241) and OD/PL (Fst: 0.129; Jost’s D: 0.238). However, the genetic similarities between the northwestern areas Burgwald-Kellerwald, Rothaargebirge, Lahn-, and Dill-Bergland resulted lower and those between the western areas Hinterlandswald and Platte resulted higher with DAPC than on the basis of Fst-/Jost’s D values.
Discussion
Limited gene flow increases genetic drift and with it the risk to loose genetic diversity (Wang and Schreiber 2001; Hartl et al. 2003; Perez-Espona et al. 2008; Frantz et al. 2012; Kropil et al. 2015; Edelhoff et al. 2020). In the long-term, this could result in increasing degrees of inbreeding (Frankham 2008; Stopher et al. 2012; Mukesh et al. 2013) and inbreeding depressions (Slate et al. 2002; Walling et al. 2011). Among the isolated AMUs, those with smaller population sizes are particularly susceptible (Whitlock 2000; Frankham et al. 2014).
This could be demonstrated in the current study for the smaller Hessian AMUs Wattenberg-Weidelsburg, Noerdlicher Vogelsberg, Krofdorfer Forst, and Gieseler Forst with an annual increase in inbreeding of 1.14 to 1.61%. In contrast, only a moderate annual inbreeding growth of 0.23 to 0.31% was calculated for the larger areas Spessart, Taunus, Reinhardswald, Meissner-Kaufunger Wald, Seulingswald, Hinterlandswald, and Burgwald-Kellerwald. Ten of the nineteen AMUs had a Ne below 100 and another two AMUs were just slightly above this threshold from which decline in fitness can be avoided in short term (Frankham et al. 2014). However, Ne from 500 to 1000 are required to maintain the evolutionary potential and a long-term adaptive potential (Frankham 1995; Franklin and Frankham 1998). These values were not achieved by any of the studied AMUs, even when subpopulations with a still assumed genetic exchange were aggregated. Red deer are polygynous and commonly display a harem mating system. However, Ne values were calculated with the Ne estimator under the assumption of a random mating system, which is not true for red deer. As a consequence, Ne values calculated under the estimation of a random mating system tend to be overestimated (Wang et al. 2016). This makes the scenario described above even worse with respect to the annual expected increase in inbreeding.
We used two approaches, a Bayesian-based approach implemented in the software package STRUCTURE and Discriminant Analysis of Principal Components (DAPC), to infer population structure of the Hessian red deer population. Unlike STRUCTURE, DAPC does not rely on any assumptions about the underlying population genetic model. According to DAPC the individuals of the 19 Hessian AMUs were divided into 15 genetic clusters. Some clusters occurred mainly or almost exclusively in individual areas. The paradigm example is cluster 9, which contained 90% of the individuals of the Odenwald, but only a few animals from other AMUs. This cluster can therefore be regarded as an indication of isolation. Besides the Odenwald, exclusive clusters were also found in the Reinhardswald (cluster 6 and 14), Knuell (cluster 2), Krofdorfer Forst (cluster 15), and Wattenberg-Weidelsburg (clusters 10 and 15). Other gene clusters occurred spatially limited in two or more adjacent areas, but included only a few animals from more distant areas, especially when separated by a fenced motorway or anthropized areas.
Whereas STRUCTURE analysis only determined four clusters on a first level, hierarchical STRUCTURE analysis detected 10 clusters on a second level that largely supports results from DAPC. Since the Dirichlet parameter α was much lower than 1 (α = 0.0531), the assumed ‘admixture’ model approaches the simpler model of ‘no admixture’. Therefore, the Hessian AMUs may be grouped into distinct subpopulations with only a few exceptions. On the basis of other studies, it can be assumed that some of the barrier effects are due to the largely fenced motorways (e.g. Wang and Schreiber 2001; Hartl et al. 2003; Frantz et al. 2012; Kropil et al. 2015). However, based on the sample density of the present study, it was not possible to separate the motorway effects from effects of other landscape elements such as widely intensive agricultural or urban areas which cannot be overcome by red deer. The motorways of the study area have been fenced since many years to avoid wildlife accidents. This interrupts old long-distance migration trails of the red deer (Herzog et al. 2020). Separating effects were also identified in regions without motorways, e.g. in a semicircle separating KF from AMUs in the north (DB, LB, BKW). In this region, there are neither fenced roads, large settlements, nor large farmlands that could function as landscape barriers. Rather, migration of red deer might be reduced by hunting in the ‘red deer–free’ areas between these AMUs.
It should be taken into account that common alleles between areas separated by a fenced motorway or other effective barriers are likely to do not only reflect the current impact of the barrier but also the historical gene flow before the barrier came into existence. Evidence for this assumption is provided by the existence of clusters (in particular clusters 3 and 5) with a widespread distribution despite the small numbers of animals belonging to them. If these clusters were able to spread beyond the barriers, the spread of those clusters which the majority of individuals belong to would be even more likely. However, it is precisely the latter clusters that show a clear spatial restriction. Unfortunately, the two effects cannot be separated. Thus, the derived degree of similarity between two areas might be overestimating the current gene flow because of the historical ‘background noise’. Thus, clusters 3 and 5 may be more indicators of historical connectivity between AMUs than evidence for current genetic connectivity. The same could also apply to other clusters, which in certain areas only occur in a few individuals.
All methods applied to uncover population structure (STRUCTURE, DAPC, Fst/Jost’s D) combined the Hessian AMUs into four regions still in genetic exchange and a number of more or less isolated areas. However, differences arise when trying to classify them qualitatively. An absolute statement on the barrier effects does not seem to be possible, because in individual cases, it can never be completely ruled out that a barrier might be overcome. Even the possibility of translocation of animals is given and has been reported anecdotally (Frantz et al. 2006).
However, even without the possibility of absolute qualitative statements, the quantitative results allow the localisation of the regions with the lowest gene flow between the Hessian AMUs.
Assuming that red deer populations in a geographically narrow region such as the study area should naturally be in a lively genetic exchange, the anthropogenic fracturing of the landscape in this region appears to have a considerable impact. These findings are in line with previous studies that identified motorways as obstacles to gene flow in red deer (Kinser and Herzog 2008; Frantz et al. 2012; Zachos et al. 2016).
In absolute terms however, the data of the Hessian population show rather favourable heterozygosity and lower Fis values in comparison with other national (Poetsch et al. 2001; Kuehn et al. 2003; Zachos et al. 2007; Edelhoff et al. 2020) and international studies (Hmwe et al. 2006a, b; Nussey et al. 2007; Nielsen et al. 2008; Sanchez-Fernandez et al. 2008; Zsolnai et al. 2009). The lowest heterozygosity and the highest F values are typically found in small islet populations and populations with longer history of isolation and low population sizes (Hmwe et al. 2006a, b; Hajji et al. 2008; Zachos and Hartl 2011; Zachos et al. 2016; Edelhoff et al. 2020). However, it must be taken into account that both measures are decisively influenced by the markers used, their number, and the sample size (Reiner et al. 2019). In the present study, more individuals (median: 59 per area; 16 microsatellites) were sampled than in similar studies (median of 25 individuals and 11 microsatellite markers; Reiner et al. 2019). As a result, more rare alleles are discovered, thus increasing heterozygosity and lowering the Fis value. Conversely, the study of Kinser and Herzog (2008) of a population in the Harz Mountains describes significantly higher genetic variability with a sample size of around 100 animals. Comparable results with regard to Fis values and allele numbers were obtained by Kuehn (2004) with samples from Graubuenden, Switzerland. However, the comparison of the results of the present study with those of Kuehn (2004) also points to obstacles between the Hessian AMUs, as the similar genetic distances in the Hessian and the Swiss study oppose considerably smaller geographical distances in Hesse, confirming a higher degree of isolation of the Hessian regions.
Low-to-moderate degree of differentiation emerged from pairwise Fst values in the Hessian population. However, Fst was originally formulated for biallelic markers, and for microsatellite markers with multiple alleles with high heterozygosity, maximum Fst is often 0.1–0.2 which is the range realised in comparisons including the Odenwald. To overcome this problem, Jost (2008) introduced ‘D’ as a new measure of differentiation. It will be 1 at complete differentiation and 0 with no differentiation. However, Jost’s D also achieves a maximum of 0.24 for an area comparison (KF to OD), which realistically no longer permits any genetic exchange due to the intervening traffic and settlement areas (Rhine-Main metropole, several fenced motorways). A comparison with Jost’s D values from Schleswig-Holstein (Edelhoff et al. 2020) showed 87% higher values for the Hessian areas. Despite the lack of absolute comparability, this at least indicates an even greater differentiation in Hesse than in the clearly isolated areas of Schleswig-Holstein that are marked by inbreeding depressions (Zachos et al. 2007). Ultimately, even with Jost’s D, no methods currently appear to be available that can solve the problems with multiple and highly variable microsatellite markers (Whitlock 2011).
Despite the difficulties with interpretation, the Fst values were cautiously compared with available values from other studies. The degree of differentiation corresponds to that of studies from Switzerland (Kuehn 2004; Fst: 0.0015–0.099), Denmark (Nielsen et al. 2008; Fst: 0.009–0.184), and Spain (Queiros et al. 2014; Fst: 0.02–0.2). Considering the insularity of the Danish populations, the isolation of the Swiss populations by mountain massifs and the distribution of the Spanish populations over a much larger area of land, the degree of differentiation of the Hessian areas can certainly be seen as an indication of the postulated isolation by legally prescribed AMUs and the landscape fragmentation. This suspicion is further confirmed by the fact that Polish populations (Niedziałkowska et al. 2012) and Scottish populations (Perez-Espona et al. 2008) on much larger areas show a much lower degree of differentiation (Fst: 0.001–0.087 [PL]; 0.015–0.022 [SC]). Significantly higher differentiation is found in red deer from southern Scandinavia (Höglund et al. 2013; Fst: 0.151–0.29), whose populations are spread over a much wider area and are divided into two subspecies between Norway and Sweden. The highest degree of differentiation is achieved in a study by Zachos et al. (2003) (Fst: 0.07–0.89), which includes large parts of southern Europe. Even if comparability between the various studies and areas is not fully ensured, the values of the Hessian AMUs nevertheless are indicative of a clear differentiation between areas in line with our starting hypothesis.
As the relatively high degrees of differentiation, compared to other red deer populations, might at least partly be explained by the motorways and their flanking regions, we recommend that AMUs be combined to larger regions and their genetic exchange enabled in order to ensure their long-term viability. In this sense, it is not sufficient to build green bridges over motorways. Rather, the paths to and from the green bridges must be ensured by biotope networking. For decades, hunters have been required by law to harvest red deer on migration in the ‘red deer–free’ areas to combat bark-stripping. The fact that this requirement is efficiently fulfilled is supported by the strong genetic differentiation of AMUs in regions without motorways and with only minor landscape fracturing (e.g. north of the Krofdorfer Forst). This harvesting also affects male red deer up to 4 years of age. At the beginning of 2019, the age threshold was raised to 5 years. This removes any basis for the possibility of gene flow and the establishment of merged gene pool between areas. Habitats must be improved and interconnected and gene flow promoted through the conservation of migratory routes used by male red deer. Currently, inappropriate hunting seasons and considerable hunting pressure lead to substantial disturbances and force red deer from the open areas into the forest, where the problem of bark-stripping damage increases. The hunting of other game species (roe deer and wild boar) in areas inaccessible to red deer and at night-time also promote the intensification of stripping damage. Habitats should be improved by establishing and improving areas inaccessible to red deer (Herzog 2019). Suitable and calmed grazing grounds can also effectively reduce damage to the forest, as can the establishment of winter feeding systems.
Promotion of genetic exchange between the AMUs might also be possible if further studies will help to identify migration corridors and possible locations for crossing-structures (e.g. green bridges) to mitigate the effects of barriers and landscape resistance on the migratory movements of red deer (Edelhoff et al. 2020).
Conclusions
A number of different evaluation strategies indicated considerable differences in the genetic diversity of the Hessian AMUs and their high level of differentiation. Areas which must be considered as largely isolated due to their extensive differentiation show Ne < 500, in many cases even < 100. This means that further losses of genetic diversity are likely in the long-term, which can threaten the viability of the local subpopulations. In order to maintain healthy red deer populations, also as a contribution to biodiversity in Hesse, current management strategies should therefore be reconsidered.
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11:155–165
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Core Team R (2017) Changes in R from version 3.4.2 to version 3.4.3. R J 9:568–570
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Earl DA, von Holdt BM (2012) A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Edelhoff H, Zachos FE, Fickel J, Epps CW, Balkenhol N (2020) Genetic analysis of red deer (Cervus elaphus) administrative management units in a human-dominated landscape. Conserv Genet 21:261–276
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRU CTU RE: a simulation study. Mol Ecol 14:2611–2620
Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66:95–107
Frankham R (2008) Inbreeding and extinction: island populations. Conserv Biol 12:665–675
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv 170:56–63
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential? Anim Conserv 1:69–70
Frantz AC, Pourtois JT, Heuertz M, Schley L, Flamand MC, Krier A, Bertouille S, Chaumant F, Burke T (2006) Genetic structure and assignment tests demonstrate illegal translocation of red deer (Cervus elaphus) into a continuous population. Mol Ecol 15:3191–3203
Frantz AC, Bertouille S, Eloy MC, Licoppe A, Chaumont F, Flamand MC (2012) Comparative landscape genetic analyses show a Belgian motorway to be a gene flow barrier for red deer (Cervus elaphus), but not wild boars (Sus scrofa). Mol Ecol 21:3445–3457
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hajji GM, Charfi-Cheikrouha F, Lorenzini R, Vigne J-D, Hartl GB, Zachos FE (2008) Phylogeography and founder effect of the endangered Corsican red deer (Cervus elaphus corsicanus). Biodivers Conserv 17:659–673
Hartl D, Clark A (1999) Principles of population genetics. Population 6:1042–1044
Hartl GB, Zachos F, Nadlinger K (2003) Genetic diversity in European red deer (Cervus elaphus L.): anthropogenic influences on natural populations. C R Biol 326:S37–S42
Herzog S (2019) Wildtiermanagement. Quelle und Meyer, Wiebelsheim, Germany
Herzog S, Schwarz UK, Leicht HJ, Krauhausen V, Voll H (2020) Lebensraumgutachten und Bewirtschaftungskonzept der Rotwildhegegemeinschaft „Krofdorfer Forst“. Rotwildhegegemeinschaft „Krofdorfer Forst“, ISBN: 978-3-00-065692-7
Hmwe SS, Zachos E, Eckert I, Lorenzini R, Fico R, Hartl GB (2006a) Conservation genetics of the endangered red deer from Sardinia and Mesola with further remarks on the phylogeography of Cervus elaphus corsicanus. Biol J Linn Soc 88:691–700
Hmwe SS, Zachos FE, Sale JB, Rose HR, Hartl GB (2006b) Genetic variability and differentiation in red deer (Cervus elaphus) from Scotland and England. J Zool 270:479–487
Höglund J, Cortazar-Chinarro M, Jarnemo A, Thulin C-G (2013) Genetic variation and structure in Scandinavian red deer (Cervus elaphus): influence of ancestry, past hunting, and restoration management. Biol J Linn Soc 109:43–53
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jost L, Archer F, Flanagan S, Gaggiotti O, Hoban S, Latch E (2018) Differentiation measures for conservation genetics. Evol Appl 11:1139–1148
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Kinser A, Herzog S (2008) Genetisches Monitoring von Rotwild in Niedersachsen–Ergebnisse einer Langzeitstudie. Deutsche Wildtierstiftung, pp 1–27
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Kropil R, Smolko P, Garaj P (2015) Home range and migration patterns of male red deer Cervus elaphus in Western Carpathians. Eur J Wildl Res 61:63–72
Kuehn R (2004) Genetic roots of the red deer (Cervus elaphus) population in eastern Switzerland. J Hered 95:136–143
Kuehn R, Schroeder W, Pirchner F, Rottmann O (2003) Genetic diversity, gene flow and drift in Bavarian red deer populations (Cervus elaphus). Conserv Genet 4:157–166
Mukesh LKS, Kumar VP, Charoo SA, Mohan N, Goyal SP, Sathyakumar S (2013) Loss of genetic diversity and inbreeding in Kashmir red deer (Cervus elaphus hanglu) of Dachigam National Park, Jammu, Kashmir, India. BMC Res Notes 6:326–331
Niedziałkowska M, Jędrzejewska B, Wójcik JM, Goodman SJ (2012) Genetic structure of red deer population in Northeastern Poland in relation to the history of human interventions. J Wildl Manag 76:1264–1276
Nielsen EK, Olesen CR, Pertoldi C, Gravlund P, Barker JSF, Mucci N, Randi E, Loeschcke V (2008) Genetic structure of the Danish red deer (Cervus elaphus). Biol J Linn Soc 95:688–701
Nussey DH, Kruuk LEB, Morris A, Clutton-Brock TH (2007) Environmental conditions in early life influence ageing rates in a wild population of red deer. Curr Biol 17:R1000–R1001
Paradis E (2010) pegas: a R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420
Perez-Espona S, Perez-Barberia FJ, Mcleodi JE, Jiggins CD, Gordon IJ, Pemberton JM (2008) Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus). Mol Ecol 17:981–996
Poetsch M, Seefeldt S, Maschke M, Lignitz E (2001) Analysis of microsatellite polymorphism in red deer, roe deer, and fallow deer--possible employment in forensic applications. Forensic Sci Int 116:1–8
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen W (2003) Documentation for STRUCTURE software: Version 2. available from: http://pritch.bsd.uchicago.edu
Queiros J, Vicente J, Boadella M, Gortazar C, Alves PC (2014) The impact of management practices and past demographic history on the genetic diversity of red deer (Cervus elaphus): an assessment of population and individual fitness. Biol J Linn Soc 111:209–233
Reiner G, Lang M, Willems H (2019) Impact of different panels of microsatellite loci, different numbers of loci, sample sizes, and gender ratios on population genetic results in red deer. Eur J Wildl Res 65:25
Reiner G, Tramberend K, Nietfeld F, Volmer K, Wurmser C, Fries R, Willems H (2020) A genome-wide scan study identifies a single nucleotide substitution in the tyrosinase gene associated with white coat colour in a red deer (Cervus elaphus) population. BMC Genet 21:14
Sanchez-Fernandez B, Soriguer R, Rico C (2008) Cross-species tests of 45 microsatellite loci isolated from different species of ungulates in the Iberian red deer (Cervus elaphus hispanicus ) to generate a multiplex panel. Mol Ecol Resour 8:1378–1381
Slate J, Van Stijn TC, Anderson RM, McEwan KM, Maqbool NJ, Mathias HC, Bixley MJ, Stevens DR, Molenaar AJ, Beever JE, Galloway SM, Tate ML (2002) A deer (subfamily Cervinae) genetic linkage map and the evolution of ruminant genomes. Genetics 160:1587–1597
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Sperlich D (1988) Populationsgenetik: Grundlagen und experimentelle Ergebnisse. Metzler-Poeschel, Stuttgart
Stopher KV, Nussey DH, Clutton-Brock TH, Guinness F, Morris A, Pemberton JM (2012) Re-mating across years and intralineage po- lygyny are associated with greater than expected levels of inbreed- ing in wild red deer. J Evol Biol 25:2457–2469
Walling CA, Nussey DH, Morris A, Clutton-Brock TH, Kruuk LEB, Pemberton JM (2011) Inbreeding depression in red deer calves. BMC Evol Biol 11:318–330
Wang M, Schreiber A (2001) The impact of habitat fragmentation and social structure on the population genetics of roe deer (Capreolus capreolus L.) in Central Europe. Heredity 86:703–715
Wang J, Santiago E, Caballero A (2016) Prediction and estimation of effective population size. Heredity 117:193–206
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whitlock MC (2000) Fixation of new alleles and the extinction of small populations: drift load, beneficial alleles, and sexual selection. Evolution 54:1855–1861
Whitlock MC (2011) G’st and D do not replace Fst. Mol Ecol 20:1083–1091
Willems H, Welte J, Hecht W, Reiner G (2016) Temporal variation of the genetic diversity of a German red deer population between 1960 and 2012. Eur J Wildl Res 62:277–284
Zachos FE, Hartl GB (2011) Phylogeography, population genetics and conservation of the European red deer Cervus elaphus. Mammal Rev 41:138–150
Zachos F, Hartl GB, Apollonio M, Reutershan T (2003) On the phylogeographic origin of the Corsian red deer (Cervus elaphus corsicanus): evidence from microsatellites and mitochondrial DNA. Mamm Biol 68:284–298
Zachos FE, Althoff C, Steynitz Y, Eckert I, Hartl GB (2007) Genetic analysis of an isolated red deer (Cervus elaphus) population showing signs of inbreeding depression. Eur J Wildl Res 53:61–67
Zachos FE, Frantz AC, Kuehn R, Bertouille S, Colyn M, Niedziakowska M, Perez-Gonzalez J, Skog A, Sprem N, Flamand MC (2016) Genetic structure and effective population sizes in European red deer (Cervus elaphus) at a continental scale: insights from microsatellite DNA. J Hered 107:318–326
Zsolnai A, Lehoczky I, Gyurmán A, Nagy J, Sugár L, Anton I, Horn P, Magyary I (2009) Development of eight-plex microsatellite PCR for parentage control in deer. Arch Anim Breed 52:143–149
Acknowledgements
The authors greatly appreciate the technical assistance of Mrs. Bettina Hopf, Ute Stoll, and Sylvia Willems. We wish to thank the chairmen and active members of the red deer conservation societies in Hesse and the ‘Landesjagdverband Hessen’ for their timely and accurate sample collection and financial support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the Hessian Ministry for the Environment, Climate Protection, Agriculture and Consumer Protection, with funds from the hunting levy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No animal experiments were performed in the present study. Samples of red deer previously shot by authorised persons during legalised hunting were used. No animals were killed specifically for the study. No living animals were sampled and no dropping antlers were sought or collected for the study.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 217 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reiner, G., Klein, C., Lang, M. et al. Human-driven genetic differentiation in a managed red deer population. Eur J Wildl Res 67, 29 (2021). https://doi.org/10.1007/s10344-021-01472-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-021-01472-8