
Abstract
The Japanese beetle, Popillia japonica, is a highly polyphagous Scarabaeidae native to Japan that colonized North America and Azores in the last century and has recently invaded Italy and Switzerland. Considering its economic impact on the horticulture and turfgrass industries, this species was ranked within the EU priority pests list in 2019. According to the EU Convention on Biological Diversity, the identification of invasion routes is a pivotal aspect in an effective management program aimed at controlling invasive alien species. To reconstruct the source of introductions of this pest, we investigated the genetic variability of P. japonica in its native and invaded areas worldwide by analyzing 9 microsatellite loci and two mitochondrial genes, COX I and CytB. In its native area, P. japonica is structured into two populations: one in the southern and another in the northern-central region of Japan. A limited area within central Japan was identified as the putative source of the North American outbreak. Moreover, the ABC inference and phylogeographic reconstruction suggest that two European populations originated from two independent introductions. The Azores Islands outbreak occurred approximately 50 years ago and originated from the southeastern region of North America (For simplicity, in this paper North America refers to Canada and the USA), while the second introduction, more recently, occurred in Italy and Switzerland and originated from northeastern region of North America.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Key message
-
Different Popillia japonica populations were identified in native area.
-
The American Popillia japonica population originated from a limited area of Japan.
-
The Azorean and European mainland populations are independent introductions from North America.
Introduction
The Japanese beetle, Popillia japonica Newman, is an invasive insect belonging to the family Scarabaeidae. Native to Japan, it is considered one of the most widespread and destructive pests of turfgrass and ornamental plants worldwide (Potter and Held 2002).
The Japanese beetle is a highly polyphagous species that can feed on more than 300 species of wild and cultivated plants as larvae and adults (EFSA et al. 2019). Due to its economic impact on the horticulture and turfgrass industries, this species was ranked within the EU priority pest list in 2019 and it is now receiving much attention from many European National Plant Protection Organizations (EU Commission Delegated Regulation 2019).
At mid-latitudes, P. japonica displays a univoltine life cycle, spending most of its life as a larva in the soil (Potter and Held 2002). In the summer, each female lays approximately 40–60 eggs, generally in the upper 7.5 cm of soil (Potter and Held 2002); the larvae develop through 3 instars and emerge as adults at the beginning of the following summer (EFSA et al. 2019). The flight period typically peaks in mid-July and adult activity terminates by the end of October (EFSA et al. 2019). Furthermore, P. japonica is not only a strong flier, able to disperse approximately 3–24 km per year (Fox 1932), but it can also relocate passively to uninfested areas through hitchhiking on nonhost commodities or vehicles (EPPO 2016).
In Japan, P. japonica occurs at high population densities in northern Honshu and Hokkaido. It is also common in Kyushu, Shikoku and southern Honshu (Fleming 1972; Klein 2008). Locally, it was not considered a pest until golf courses were damaged because of larval activity. The first report of P. japonica outside of Japan occurred in 1916 when it was first detected in southern New Jersey, USA (Fleming 1976). In the following years, it colonized the entire eastern USA and progressively expanded to the Midwest. With the exception of Florida, which most likely has an unsuitable climate, the Japanese beetle became established in all states east of the Mississippi River by 1998 (Potter and Held 2002). Only sporadic occurrences are currently reported from the western USA, where several eradication campaigns have been carried out using chemical insecticides (Clair and Kramer 1989; Hungate et al. 2016). In Canada, P. japonica was first detected in 1938 and is now present in the eastern provinces of Ontario, Quebec, Nova Scotia, New Brunswick, and Prince Edward Island (CFIA 2009). In 2017, an infestation was discovered in Vancouver, British Columbia but this population is currently being eradicated (EPPO 2018). Outside of North America, P. japonica reached the Azores (Portugal) in the 70s and is now established on several islands in this region, although at different population densities (Potter and Held 2002). The first outbreak in mainland Europe occurred in Ticino Natural Regional Park, northern Italy, in 2014 (Pavesi 2014). This area is located between the Piedmont and Lombardy regions in an area where two airports, one commercial and one military, are present. In 2017, P. japonica was also reported from southern Switzerland, possibly as an expansion of the Italian infestation (EPPO 2017). Scattered reports of P. japonica in the Korean Peninsula, China and India may on the other hand have arisen from misidentifications (Potter and Held 2002).
Losses due to P. japonica feeding on wild and cultivated plants can be substantial. Although it is uncertain how much economic damage Japanese beetles cause in Europe, it is estimated that more than 450 million dollars are allocated annually to manage the spread of this pest and to replace damaged plants in the US (Potter and Held 2002). In Italy, several measures are being employed to limit the distribution of Japanese beetles. Natural and commercial entomopathogenic nematodes and fungi have been tested and applied to infested soils (Benvenuti et al. 2019; Marianelli et al. 2018; Mazza et al. 2017; Paoli et al. 2017; Torrini et al. 2020) to control larvae. Attract and kill devices, made of long-lasting insecticide-treated nets with a double lure, along with “attract and infect” devices equipped with entomopathogenic fungi and also a double lure, have been developed and deployed in infested areas to control adult populations (Marianelli et al. 2019; Regione Piemonte 2021). Even with these control tactics, P. japonica has invaded as much as 16,000 square kilometers in northern Italy and in the neighboring Swiss Ticino Valley since 2014 (Mori et al. 2022).
The experience of the recent decades suggests that knowledge of the genetic structure and the routes of introduction of an invasive species can assist in developing effective monitoring programs and limit additional introductions (Nardi et al. 2005; Roderick and Navajas 2003). Moreover, novel approaches are being developed that use molecular targets for the genetic control of pests, further underlining the importance of genetic data and information on the geographic structuring of genetic variability (Scott et al. 2020).
To determine the current distribution, population structure and relationships of P. japonica, our research investigated the genetic variability of this insect in Japan and in invaded areas worldwide by reconstructing the phylogeographic history using ABC inference and haplotypic networks. Analyses were carried out with data obtained from 9 microsatellite loci and two mitochondrial genes, COI and CytB.
Materials and methods
Sampling and DNA extraction
Adult and larval P. japonica were collected from several areas in its current global distribution. More than 215 individuals were collected, and the sampling coordinates, date of collection, and name of collector were recorded for each specimen (Supplementary Table 1). Following the manufacturer’s recommended protocol, DNA was extracted from adult thoracic muscles and the last three larval metamers, using the Blood and Tissue DNA Mini Kit (QIAGEN). The final elution step was performed in 100 μl of AE buffer supplied in the commercial kit.
Microsatellite development
Low coverage sequencing of the P. japonica genome was performed at BioFab Research s.r.l. (Rome, Italy) to produce a total of 2.31 million contigs (from 78 bp to 978 Kbp) that encompass 1.57 Gbp of DNA sequences (data not shown). Automatic extraction of microsatellite loci was performed to identify 1620 regions that included sequences (di-, tri- and tetranucleotide) longer than 10 repetitions. Based on the initial observation that different contigs share regions of sequence similarity and that ancillary repeats, simple sequences and homopolymer runs are frequently observed in flanking regions, candidate loci were subject to a filtering step before primer design.
Blastn ver. 2.6.0 + , function makeblastdb (Camacho et al. 2009), was used to compare the 1620 regions (repeat and 200 bp flanking sequences on each side) against the genomic assembly under default settings that, in terms of stringency, roughly correspond to a match of 100 bp with 90% similarity. Regions that produced more than one match against the genomic assembly were discarded.
The occurrence of sequences characterized by low complexity in the flanking regions, which could complicate primer design and sizing, was evaluated using a modified Lempel–Ziv algorithm (Orlov and Potapov 2004) (http://wwwmgs.bionet.nsc.ru/mgs/programs/lzcomposer/index.html). This measure, developed for the study of noncoding genomic regions, is stable for complex sequences, but displays a significant drop in the presence of dispersed repeats, palindromes, homopolymer runs and other sequences of low complexity. Sequence complexity was evaluated in a window of 40 bp with 10 bp increments over 200 bp flanking regions on each side. Regions with at least one segment of 40 bp with an associated complexity lower than 15 were discarded. The threshold was adjusted based on visual observation of the results. Furthermore, only sequences characterized by perfect di- and trinucleotide repeats, recorded in contigs of length > 3000 bp, with an A + T content < 70% and devoid of homopolymer runs > 5 bp in the 200 bp flanking the repeat were retained. The resulting 71 regions were visually inspected and 20 were selected for primer design and testing. Primers were designed using the primer-blast (Ye et al. 2012) implementation of Primer3 (Untergasser et al. 2012).
Optimization of candidate loci
Twenty candidate primer pairs were used for testing and optimization of amplification conditions. Amplifications were performed using a GoTaq G2 Flexi DNA Polymerase (Promega, Madison, WI, USA) following the manufacturer’s specifications, with cycle 95 °C 2 m; 95 °C 1 m, 50–58 °C 1 m, 10 s 72 °C 1 m 30 s × 35 cycles; 72 °C 10 m, 4 °C 10 m on a GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA, USA). Following optimization of PCR conditions, fragments that produced clear amplicons were cloned in a pGEM-T Easy Vector System (Promega, Madison, WI, USA) and sequenced to confirm locus specificity. Nine loci that produced the most consistent and clean amplifications were selected for genotyping. Microsatellite sequences have been deposited in GenBank under accessions OL802935/OL802944.
Genotyping
Genotyping was performed on 16 individuals for each of the 7 sampling areas (112 individuals in total). Sampling areas were defined according to geographical features: Japan was subdivided into three regions (south, east and central-west areas) based on the archipelago geography, the USA and Canada into two regions (northeast and southeast), and Azores and Italy were considered as distinct areas. All forward primers (BioFab Research s.r.l., Rome, Italy) were marked with 6-FAM dye. Amplifications were performed as above, with annealing temperatures as indicated in Supplementary Table 1.
Genotyping was performed at BioFab Research s.r.l. (Rome, Italy) on an ABI3730 DNA Analyzer (ThermoFisher Scientific, Waltham, MA, USA) using a 1:200 dilution of PCR products. The LIZ conjugated ladder Gene Scan 500 (ThermoFisher Scientific, Waltham, MAS, USA) was used for sizing, with the exclusion of the 250 bp band following the manufacturer’s instructions. Scoring of peaks was performed using Geneious (v.11.1.3, Biomatters, Auckland, New Zealand). The region between ladder bands 50–500 bp was evaluated and peaks were called using the third-order least squares approximation. Bins were automatically inferred. Automatically detected peaks were manually revised to account for locus specific idiosyncrasies (e.g. tendency to form stagger bands) and read into a spreadsheet. One locus (F) was excluded because of the frequent presence of smearing instead of amplification bands. The final dataset is available as Supplementary Data.
Microsatellite analysis
Presence of the null allele was evaluated through the Hardy–Weinberg test for heterozygote deficiency (Guo and Thompson 1992; Rousset and Raymond 1995) in each population, while its frequency was estimated according to Dempster et al. (1977) using FreeNA software (Chapuis and Estoup 2007; Chapuis et al. 2008) and corrected for possibly failed amplifications (beta parameter) using ML-Null software (Kalinowsky and Taper 2006). Genetic distance and Fst were calculated with ML-Null software considering the presence of null alleles and beta correction.
Basic locus statistics were calculated using GenePop v. 4.7 (Raymond and Rousset 2001; Rousset 2008). Clustering of individuals into putative populations was obtained in Structure v. 2.3.4 (Falush et al. 2003; Pritchard et al. 2000) using the admixture and correlated model, as well as the locprior option (Hubisz et al. 2009) as suggested in the software documentation for small sized datasets.
Fifteen independent runs were conducted for each possible value of K (1 to number of sampling areas in different analyses) with a burn-in values of 50.000 and 100.000 MCMC generations after burn-in. The optimal K was selected using the Evanno method (Evanno et al. 2005) as implemented in Structure Harvester v. 0.6.94 (Earl and vonHoldt 2012). Among multiple replicates for a given K, the one with the highest likelihood was used for visualization in Structure Plot v. 2.0 (Ramasamy et al. 2014). The analysis was initially conducted on the full dataset. To deploy knowledge on the sequences of introduction/establishments in different areas, subsets of the data pertinent to specific phases/events of introduction were also analyzed separately (Japan only, Japan + USA, USA + Azores + Italy).
Principal coordinates analysis (PCoA) was conducted on microsatellite data to identify genetic ancestry differences between P. japonica populations. Genetic data on distance between populations were considered including the presence of the null allele. Computation was performed with GenAlEx 6.5 add-in for Microsoft Excel spreadsheet (Peakall and Smouse 2012).
Amplification of mitochondrial targets, sequencing and basic data analysis
Fragments of the mitochondrial genes COX I and CytB were amplified using the LCO1490/HCO2198 primers (Folmer et al. 1994) and CP1/CB-N-11367 primers (Harry et al. 1998; Simon et al. 1994), respectively (Supplementary Table 1). Amplifications were performed in a total volume of 50 μl containing 25 μl of HS DreamTaq Master mix (2X) (Thermo Scientific) 0.6 mM of each primer and 10 ng of template DNA. The thermal protocol used for the amplifications was 94 °C 3 m (for HS activation); 95 °C 30 s, 40–45 °C 30 s, 72 °C 45 s × 45 cycles; 72 °C 5 m. PCR products were checked on a 2.0% agarose gel, purified with QIAquick PCR purification kit (QIAGEN), and sequenced in both directions at BioFab Research s.r.l. (Rome, Italy). Sequences were assembled using Geneious (v.11.1.3, Biomatters, Auckland, New Zealand). Samples with incomplete or low-quality sequences in one or both mitochondrial loci were discarded in the subsequent analyses.
Alignments of the resulting 187 sequences were performed with ClustalOmega and manually inspected. Haplotype diversity (Hd), the average number of nucleotide differences (k), nucleotide diversity (π), Tajima’s D (Tajima 1989), and Fu’s Fs (Fu 1997) neutrality indices were calculated using Arlequin v. 3.5.2.2 (Excoffier and Lischer 2010) for both loci.
Phylogeographic analyses
Maximum likelihood (ML) analyses were performed on individual loci and the concatenated dataset to evaluate the congruence between the two loci. The appropriate substitution matrices were chosen based on JModelTest v. 2.1.10 considering the corrected Akaike information criterion. Trees were computed using MEGA-X software adopting TN93 as the substitution matrix in all three cases (COI, CytB, and concatenated loci) and a moderate branch swapping algorithm. A bootstrap test with 1000 pseudoreplicates was conducted.
Haplotype networks were reconstructed using the TCS algorithm (Clement et al. 2002) in PopArt (Leigh and Bryant 2015) for three COX I–CytB combined datasets: all samples, Japanese samples and introduced-areas samples.
ABC analysis was performed to compare different colonization scenarios using DIYABC v.2.1.0 (Cornuet et al. 2014). The 9 microsatellite and 2 mitochondrial loci were used as initial data. All possible scenarios that hypothesize Japan as the native area of the species and the USA as the first colonization area were tested, and Japan was subdivided into two areas (north-central Japan and south Japan) based on geography to test the origin of the US outbreak. Six possible scenarios fit the chronological outbreak events that occurred in the last century, and they were assumed at the same probability as priors. The number of generations was estimated by assigning 1 generation/year as the demographical parameter (Fleming 1972; Potter and Held 2002) and the first official outbreak report as the starting colonization time in a new area. Molecular data were divided into two groups; the mutation model of mitochondrial data was set to TN93, and the upper limit of the mean mutation rate was adjusted to 10−8 while microsatellite priors were set as the default. Each scenario was tested in 1 × 106 simulated datasets, and the population range size was adjusted from 104 to 108. The posterior probabilities of the competing scenarios were computed using direct and polychotomous logistic regressions on the 1.0% of simulated data closest to the observed dataset, and the most likely scenario was considered the one characterized by the highest significant probability value. The posterior distributions of demographic parameters were estimated under the chosen scenario on the 1% closest simulated data sets.
The Bayesian reconstruction of phylogeographical relationships between samples was performed using *BEAST2 v. 2.6.4 software (Bouckaert et al. 2014) plus bModelTest v. 1.2.1 (Bouckaert and Drummond 2017), model-selection and GEO_SPHERE v. 1.3.0 (Bouckaert 2016) as additional packages. Data were arranged in three partitions, two containing nucleotide sequences and the third containing geographical coordinates of the sampling area. Samples were arranged into 39 different OTUs named from the geographical regions where specimens came from. The most suitable substitution matrix for each nucleotide partition was TN93 and the coalescent exponential growth was applied as a demographic tree prior to a strict molecular clock model. MCMC was run for one billion generations, sampling every 5000 generations and discarding 10% as burn-in.
Results
Dataset evaluation and basic statistics
From the initial 215 samples, we successfully amplified and completely sequenced of both mitochondrial loci in 183 Popillia japonica, 3 Popillia quadriguttata and 1 Popillia mutans specimens. Sequences were submitted to GenBank; accession numbers are reported in Supplementary Table 1.
The analyzed regions of COX I and CytB mitochondrial genes of Japanese beetles were 657 and 744 nucleotides long, respectively, and did not include any gaps or missing data. Genetic indices and neutrality tests calculated for both loci are summarized in Table 1. Considering the species in the whole area of distribution, Tajima’s D and Fu’s Fs values for the COX I and CytB loci were negative (only COX I Tajima’s D was not significant) confirming the recent Japanese beetle general expansion. The Italian and Azorean populations of P. japonica had a negative and significant Tajima’s D that identified a population expansion but with a balanced number of haplotypes (nonsignificant Fu’s Fs). In North America and in the southern Japan, the populations can be considered constant in size with a balanced number of haplotypes, while in the northern and in the central areas of Japan the number of haplotypes was significantly higher than expected (negative and significant Fu’s Fs) with a nonsignificant variation in population size.
Genotyping produced clearly identifiable allele data for all the 9 microsatellite loci targeted, with a minimal fraction of missing data (3%). Peak identification was straightforward and required minimal manual curation from the automated Geneious scoring. Loci were generally quite variable, with 6 to 30 alleles per locus overall and values of expected heterozygosity (He, average across populations) from 0.34 to 0.86 (Supplementary Table 1). He was higher in Japan (0.78 ± 0.01; mean ± s.d.) than in invaded areas (0.65 ± 0.06). The Hardy–Weinberg exact test, conducted by locus and by population, rejected equilibrium (p < 0.01) in all but two instances (i.e. loci A, B).
The Hardy–Weinberg exact test for heterozygote deficiency confirmed the presence of null allele in all loci but A and B (Supplementary Fig. 2A). The loci with the highest degree of differentiation were locus C and locus G with Fst values of 0.20 and 0.15, respectively (Supplementary Fig. 2B). Fixation index values across populations identified a considerable degree of differentiation of southern Japanese population with the others; moreover, a relevant Fst values were also found between the Azorean and Italian populations (Supplementary Fig. 2B). These populations have a genetic distance closer to northern American populations than Japanese populations (Supplementary Fig. 2C).
Microsatellite data
Analysis of the full dataset (7 sampling areas) suggested K = 2 as the optimal number of clusters, with a secondary signal (visual observation of likelihood plateau) at K = 4. If two clusters are assumed (Fig. 1A; Supplementary Fig. 3A), southern Japan is occupied almost exclusively by one cluster (yellow in Fig. 1A) while eastern and central-western Japan appear to be characterized by a mixture of both clusters in similar proportions. All invading populations belong to the latter cluster (orange), with a minimal reminiscence of the former in southeast USA. Partitioning into four clusters (Fig. 1B; Supplementary Fig. 3B) led to the identification of (a) a first group (yellow) almost exclusive in southern Japan; (b) intermixing of three groups in east and central-west Japan (yellow, azure, green), with a dominance of the former in central-west Japan; (c) intermixing of the latter two groups (azure, purple, not yellow) in the USA and their separation in the Azores and Italy, which appear populated by one different group each.

Structure analyses for the full microsatellite dataset assuming K = 2 (A) and K = 4 (B)
Analysis of the Japanese dataset (3 sampling areas) suggested K = 2 as the optimal number of clusters. One cluster corresponds unequivocally to southern Japan and the other to eastern and central-western Japan (Supplementary Fig. 3C). Analysis of the Japan + USA dataset (5 sampling areas) suggested K = 2 as the optimal number of clusters. One is identified in southern Japan while a combination between this and a second cluster characterizes eastern and central-western Japan. Only the latter is present in the USA (Supplementary Fig. 3D).
Analysis of the USA + Azores + Italy dataset (4 sampling areas) suggested K = 2 as the optimal number of clusters, with a secondary signal at K = 3. Assuming 2 clusters, one characterizes the USA and Azores, whereas the second cluster is observed in Italy (Supplementary Fig. 3E). Assuming 3 clusters, each of the 3 sampling areas is characterized by the dominance of one different cluster, with a higher level of intermixing in the USA than in the Azores and Italy. While differences in the less frequent groups are limited and not readily discernible, the cluster characterizing the Azores is present in the USA and absent in Italy (Supplementary Fig. 3F).
Principal component analysis of 9 loci of microsatellite data (Fig. 2A) showed a distribution of the southern Japanese population of P. japonica with respect to the other populations of the same geographical origin. Furthermore, the Italian and Azorean populations also showed a significant genetic distance. Analysis based on mean intra-population genetic distance (Fig. 2B) revealed the two major PCoA components accounting for the 87,02% of the total variation, while five factors were required to describe the total variation in the dataset (Eigen value > 1).

PCoA of 9 loci of microsatellite data A on individuals and B on populations. The presence of null alleles was considered in genetic distances between populations
Mitochondrial data
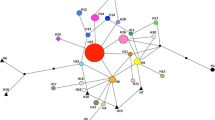
The genetic trees based on the two mitochondrial genes COX I and CytB obtained using ML were congruent and did not have topological conflicts, but displayed weak support for the regional subdivision of Japanese samples and, therefore, did not have sufficient resolution to determine the origin of samples from non-native areas (Supplementary Figs. 4 and 5). The TCS network constructed with COX I–CytB combined data obtained from the entire distribution of P. japonica included 84 nodes, but only 65 were vouchered haplotypes, with an average distance between nodes of 2.34 units with mode 1, and only a few nodes were located very far from the main core of the network. The majority of haplotypes (n = 53) occur in Japan, but only 3 of these are also present in the colonized areas while 10 haplotypes only occur outside the native area (Fig. 3; see Supplementary Table 1 for full location information).

Popillia japonica haplotypic network considering all collected samples. Hatch marks represent the number of substitutions between haplotypes in the mtCOX I and mtCytB concatenated dataset. The histogram shows the mean number of substitutions in the haplotypic network
Two separate TCS networks using haplotypes found in samples collected in native and nonnative areas were computed to relate the haplotypic data to geographical regions of origin. The TCS network obtained only with Japanese samples connects 53 vouchered haplotypes with an average distance of 2.79 units (Fig. 4).

Popillia japonica haplotypic network considering only samples collected in the native area and its subdivision into groups. Hatch marks represent the number of substitutions between haplotypes in the mtCOX I and mtCytB concatenated dataset. The right panel shows the geographical location and the relative abundance of haplotype groups
Haplotype groups are geographically restricted to specific Japanese regions, e. g.: group IX was found only in the southern Japanese islands of Kyushu and Tsushima, while groups I and III were found in the center of Honshu. Other groups have a wider distribution such as IV and VII, which were observed from the center of Honshu to Hokkaido. Although some groups were widespread across Japan, no single haplotype or group was observed throughout the archipelago.
The TCS network obtained from the introduced area displayed less variability compared to the Japanese network (n = 13), with only 8 haplotypes observed exclusively in North America (USA and Canada), two haplotypes among Azorean and Italian beetles, and one in the Swiss population (Fig. 5A). Only three North American haplotypes were found in Japan (Hap_34, Hap_38, and Hap_48) where they were located in a geographic region that comprises the Gunma, Ibaraki, and Tochigi prefectures. The most common and widespread haplotype in the North American continent was Hap_38 while Hap_48 was found only in a limited region in the USA between New Hampshire and Maine. Only two haplotypes (Hap_38 and Hap_59) were identified in the Azores Islands.. The same haplotypes of Azores were identified in the Italian outbreak of P. japonica, although with substantially different frequencies, while only Hap_59 was found in samples from Switzerland.

A Popillia japonica haplotypic network considering samples collected in the introduced areas. Hatch marks represent the number of substitutions between haplotypes in the mtCOX I and mtCytB concatenated dataset. The right panel shows the geographical location of haplotype groups in Japan (B), North America (C), Azores (D), Italy and Switzerland (E). The color code was the same as that adopted in Fig. 3. Arrows represent the preferential migration direction of the two North American populations of Japanese beetles: orange color east to west, red from north to south
The phylogeographical reconstruction based on multipartition data (COX I, CytB, and geographical coordinates of sampling sites) obtained by Bayesian inference supports the subdivisions previously found between mitochondrial haplotypes and geographical coordinates. Moreover, the Bayesian tree was able to partially resolve the topology of nonnative areas (Supplementary Fig. 6A and 6B).
Haplotypic groups previously identified in the native-area network were generally monophyletic, except for Group V which was definitively paraphyletic, and Groups II and III, which were not well resolved at a posterior probability threshold of 0.85. Specimens collected outside the native range occurred in two clades that were substantially superimposable on Group II and Group III. In North America, specimens belonging to Group III were found in the US Midwest, eastern USA and eastern Canada, while Group II specimens were collected from the US Midwest, eastern USA and western North America. The recent introductions in British Columbia, Oregon, Idaho and Colorado belonged to Group II.
To test the hypothesis of a bridgehead scenario where double and independent introductions occurred, a subset of samples characterized with both mitochondrial and microsatellite data, were chosen to perform an ABC analysis. The ABC inference identifies the hypothesis of two independent introductions for European outbreaks from the US as the most likely scenario in both model choice approaches, supporting the conclusion obtained from the complete datasets.
The most likely historical model identified by the ABC approach identified the origins of the North American outbreak of P. japonica as an introduction from the north-central region of Japan, while the Azorean and Italian populations were independent introductions from North America. The six possible models were tested with both direct and logistic regression (Fig. 6B), and the confidence in scenario choice was 0.747 and 0.735, respectively. Comparison of datasets generated from priors and posterior distribution of Scenario 2 parameters (Fig. 6C) revealed a higher number of observed mitochondrial alleles and segregating alleles against the predicted alleles only in north-central Japan, A possible explanation of this discrepancy will be discussed in the subsequent section.

ABC analysis. A all the scenarios tested. Scenario number 2 was identified as the most likely hypothesis. B choice of the most likely scenario using direct and logistic decision criteria. C estimated and observed parameters considered in the ABC reconstruction with the proportion of the simulated dataset with a value below the observed data for the chosen scenario number 2
Discussion
Popillia japonica was a fairly neglected phytophagous species until it came to the limelight following its invasion of North America and the Azores in the last century and Italy more recently. A rich scientific literature has since accumulated on outbreaks and their spread, as well as on the phenology of the species in different ecological settings (Fleming 1972; Potter and Held 2002; CFIA 2017; USDA 2022). The increasing knowledge about the biology of this species, including its ecological traits and a list of its most suitable host plants, has made it possible to carry out new control approaches with targeted measures (Sipolski et al. 2019; Althoff and Rice 2022). Moreover, as pointed out in the decision VI/23 (paragraph 14) of the EU Convention on Biological Diversity (SCBD) (EU 2002), the identification of the invasion route is a pivotal aspect in an effective management program aimed at controlling invasive alien species (IAS). Knowledge about the geographical origin of an IAS species facilitates strategic planning to control or prevent invasions in a more comprehensive framework, improves phytosanitary inspection protocols, identifies environmental and evolutionary factors potentially involved in biological invasions (Estoup and Guillemaud 2010), and halts gene flow between populations to prevent or reduce the selection of genetic traits in introduced populations that promote invasiveness (Facon et al. 2006; Schierenbeck and Aïnouche 2006). These aspects acquire a particular relevance when the involved species are well known for their damaging potential. There is consensus about human-mediated introductions of the species from its native range in Japan to the North America, to the Azores Islands and to the mainland Europe (EPPO 2020), yet a reconstruction of the historical invasion routes of this species using a phylogeographic approach has been missing until now.
Genetic analyses of Japanese samples of P. japonica identified two different lineages in the native area, the first in Kyushu and Tsushima and the second across Shikoku, Honshu and Hokkaido. This separation between southern and north-central Japan was jointly supported both by microsatellites and by mitochondrial data, with the identification of two different genetic clusters in the two areas. Haplotype group IX, characterizing the southern islands, is separated by 13 substitutions from Group VIII (Shikoku) and 33 substitutions from the core of the network (Honshu and Hokkaido). This differentiation may have been promoted by the presence of the Seto Inland Sea, which could have acted as a natural barrier to gene flow separating Kyushu from Shikoku and Honshu over historical times. A parallel differentiation was, on the other hand, not observed between Shikoku and Honshu, with samples clustering in the same group based on microsatellite data and extensive sharing of haplotypes belonging to Group V across the two islands. Whereas the entire Honshu was recovered as a single assemblage based on microsatellite data, mitochondrial haplotypes recovered additional structure in the area. The highest haplotypic richness was observed in central Honshu, suggesting that this location may be the origin of this species, with other Japanese islands having been subsequently colonized through a process of range expansion that occurred at some point in the past. Haplotypic data from samples collected in the northern part of Japan (Hokkaido and Akita Prefecture in Honshu) were in turn characterized by Group IV and Group VII haplotypes, further supporting an old expansion process from the central region of Honshu to Hokkaido. Taken together, these observations suggest an old process of differentiation of P. japonica in its native area, with an origin in Honshu and a secondary differentiation in the northern and southern islands. This scenario is also supported by PCoA analysis, where the southern Japanese population is the only subdivision that clearly differentiates from the other Japanese samples (Fig. 2A). The ABC reconstruction estimated a divergence time of approximately 38.000 years between Japanese populations.
In the light of the biological features and the possibility of spreading of P. japonica, its presence in North America is undoubtedly associated with human activities and trade that took place in the last century. Microsatellite analysis and ABC reconstruction identified the origin of the North American outbreak from the eastern and central-western Japan populations. Based on the haplotypic network, the presumptive geographic origin of the invading population could be narrowed down further to central-eastern Honshu. Out of the 13 haplotypes found in North America, only 3 were identified in Japan, namely Hap_34, Hap_38, and Hap_48, with the first two being the most frequent overall in North America and having spread and differentiated to produce a series of ancillary haplotypes with a spotted geographical distribution It is therefore possible to hypothesize that Hap_34 and Hap_38 dominated the original introduction, with other satellite haplotypes being generated by casual mutation since then. These two most frequent North American haplotypes were identified only in a limited geographical region of Japan, in the center of Honshu Island between the Ibaraki, Tochigi, and Gunma Prefectures. As such, this geographical region can be considered as the most likely origin of the invasive specimens associated with the first North American outbreak of P. japonica.
An alternative hypothesis is that a larger number of mitochondrial maternal lineages characterized the first introduction of P. japonica in North America but only two of them (Hap_34 and Hap_38) were widely spread in that area. The other introduced haplotypes that occur in a spotted distribution in North America, could be present in the same Japanese area as most abundant ones between Ibaraki, Tochigi, and Gunma Prefectures but with a very low frequency. Their rarity made sampling of these haplotypes an occasional occurrence.
Being present in Japan in the same region (Nagano Prefecture), Hap_48 may be part of the original introduction, having followed the same route hypothesized for Hap_34 and Hap_38. Nevertheless, the observation that it is found at low frequency in North America with a geographical distribution restricted to Maine (USA), gives credence to the alternative possibility that it may be the outcome of an independent outbreak that occurred more recently in a limited geographical area in the US.
Microsatellite analysis of the Azorean and Italian samples allowed only a weak definition of the presumptive area where European outbreaks originated, as the USA is characterized by a substantial intermixing of the groups that are then observed in isolation in the Azores and Italy. This consideration is also supported by the PcoA analysis (Fig. 2A), where no clear differentiation could be found in the northeastern region of North America with respect to southeastern regions.
Based on mitochondrial data, out of the two haplotypes found in the Azorean Japanese beetles, Hap_38 is common in the US, while Hap_59 is observed in Colorado, New Jersey, and Canada’s British Columbia. This latter differs from the more common Hap_38 by only 2 substitutions, with the intermediate haplotype Hap_62 identified in samples from North Carolina (USA). A possible origin of the P. Japonica outbreak in the Azores could therefore be tracked to the US east coast. In fact, following from the idea of a continuum spread during the North America colonization (Allsopp 1996; Bai et al. 2017 USDA 2020; CERIS 2020; Althoff and Rice 2022), the first invaders characterized by Hap34 and Hap_38 could have spread in the south where Hap_62 and the subsequent Hap_59 were generated during the 50 years of colonization. The high number of individuals reached in the first years of spreading in rural areas in North America (Davis 1920) could have played a relevant role in the fixation of those haplotypes in the local gene pool.
The initial invasive population of P. japonica established in North America could be the source of additional outbreak via secondary introduction in the Azores that occurred more than 50 years ago. The traits drawn by this scenario were defined by Lombaert and et al. (2010) as “bridgehead effect,” which has been demonstrated as a dynamic of invasion for other insect pests, such as Coptotermes formosanus (Blumenfeld et al 2021). A bridgehead scenario can also explain why the location of the first outbreak in Terceira Island (Azores) was found near the US Airforce base “Lajes” (Martins and Simoes 1985) and the low haplotypic variability observed in the Azores.
The Italian outbreak is characterized by the same haplotypes of the Azorean population, nevertheless their haplotypic frequencies are totally different. As such, and even allowing some shift in genetic frequencies associated with low population number during the invasion, it is unlikely that the Italian outbreak originated from the Azores. This conclusion is further supported by microsatellite analysis, in which the Azores and Italy are tipically characterized by different genetic clusters for K > 2, and confirmed by PcoA and ABC reconstruction, in which outbreaks in Azores and Italy were identified as independent introductions. Hap_59 was identified in three location in North America, including New Jersey, which appear to be the most likely source of this outbreak, since Colorado and British Columbia are of recent detections (2018 and 2017, respectively) and the pest is not considered locally established in these two locations (CFIA 2020; EPPO 2018).
Circumstantial evidence suggests a possible correlation between air traffic and P. japonica, particularly the position of the initial Italian outbreak in an area between Milano Malpensa International Airport and Cameri military airport (EPPO 2014). Moreover, Hap_59 and Hap_38 haplotypes are present in sympatry only in New Jersey, where the Newark International Airport is located, which is historically the largest airport in the US and currently part of the largest airport system worldwide. Altogether these observations allow us to hypothesize a possible origin of the Italian specimens from the New Jersey area mediated through accidental transport by plane. This trajectory is a common scenario for human-mediated dispersal of alien species as described by Banks et al. (2015), and its probability is increased due to the involvement of international hubs with a high volume of traffic (Floerl et al. 2009). Plane-mediated transport is a known vector of alien pests as reported in many inspection report surveys (Liebold et al. 2006). Flights were suspected to be the cause of repeated detection of Ceratitis capitata in California in years following eradication campaigns (Liebhold et al. 2006) and airplanes are the most suspected vector of introduction of Diabrotica virgifera virgifera in Europe (EPPO 2012).
The Swiss outbreak is monomorphic for Hap_59, which is the dominant haplotype in Italy. This occurrence, together with the geographical continuity between the two sites, strongly suggests that the spread of Italian P. japonica in the neighboring Ticino area of Switzerland was the origin of the Swiss outbreak.
A consideration should be made on the number of the mitochondrial haplotypes in northern-central Japan. Even if a possible explanation of the discrepancy between simulated and observed haplotypes found in ABC reconstruction can be due to the inaccuracy of mean mutation rate estimation based only on one thousand years of mutation events to describe longer time-scale period events, however the number of haplotypes observed in the northern and central regions of Japan was found to be significantly higher than expected also considering the Fu’s Fs parameters. A possible hypothesis to explain this excess of alleles is the existence of a different demographic history for northern-central populations of P. japonica than for southern ones. Another possible explanation is the different density of golf courses in the center of Japan than in the southern regions (Saito 2009). A widespread presence of chemically treated grass fields, such as golf courses, can act as a large-scale attract-and-kill trap that reduces population density by applying a constant selective pressure on the local population of P. japonica.
The occurrence of the bridgehead effect in the spreading process of P. japonica is also confirmed with the ABC analysis, because the “all independent introduction from native area” scenario has the lowest statistical probability; in contrast higher probability values were scored by the two models that hypothesize European outbreaks to be related to the North America population. This conclusion is also supported in the full mitochondrial and microsatellite dataset, where only a limited group of recurrent alleles were found in nonnative areas.
To add the information on distances between sampling areas, we performed a Bayesian phylogeographical reconstruction using both geographical coordinates and COX I and CytB mitochondrial data. Microsatellite data were not included in this analysis due to the long and intensive computation required. In the native range the previously identified groups were monophyletic with the exception of Group V whose paraphyly can be due to a spotted geographical distribution (Supplementary Fig. 6B). The outbreaks discovered in the western region of North America were originated only from specimens belonging to Group II, as a result of introductions from the east to west, while the eastern Canadian populations (originated by Group III) arose due to the establishment of individuals that had spread from southern locations. Even if it is possible, different migration patterns occurred between the two North American populations of P. japonica. The migration could also be explained by the existence of two other human-mediated spreading pathways of Japanese beetles through the USA and Canada, but these are still unidentified.
Conclusions
The Japanese beetle is an important pest, noted in the alert list in several countries around the world due to its polyphagy; it has the potential to create major damage to cultivated and wild plants and proclivity for long-range successful introductions. In this work, we have described older differentiation of the species in its native area with sufficient resolution to track back the origin of the North American P. japonica outbreak to a limited region in the center of Honshu. Our reconstruction found support for a distinct origin of European P. japonica, with the Azorean outbreak presumably originating from the southeastern USA and the Italian-Swiss outbreak independently introduced from the northeastern USA. Our results also support the hypothesis that airports are high-risk entry points for newer accidental introductions of this species. As such, the improvement of methods to evaluate the presence of P. japonica adults and larvae in traded plants and commodities may be warranted.
Author contributions
AS conceived the experiments; AS, FP, FN, KS, TK, AC and LM planned sampling; AS, NF and II performed laboratory work; AS and FN analyzed the data; AS, FP, NF, II, TK, AC and LM wrote the manuscript; GB, PFR and LM provided funding for the research; and TK revised the English language. All authors read and approved the manuscript.
Data availability
All sequence data are available in GenBank database, and microsatellite data are available in the Supporting materials.
References
Althoff ER, Rice KB (2022) Japanese beetle (Coleoptera: Scarabaeidae) invasion of North America: history, ecology, and management. J Integr Pest Manag 13:1–11. https://doi.org/10.1093/jipm/pmab043
Allsopp P (1996) Japanese Beetle, Popillia japonica Newman (Coleoptera: Scarabaeidae): Rate of Movement and Potential Distribution of an Immigrant Species. The Coleopterists Bull 50(1):81–95. https://www.jstor.org/stable/4009259
Bai BB, Burfitt CE, Hedstrom CS (2017) Japanese beetle in Oregon. https://www.oregon.gov/ODA/shared/Documents/Publications/IPPM/JapaneseBeetleInOR.pdf
Banks NC, Paini DR, Bayliss KL, Hodda M (2015) The role of global trade and transport network topology in the human-mediated dispersal of alien species. Ecol Lett 18:188–199. https://doi.org/10.1111/ele.12397
Benvenuti C, Barzanti GP, Marianelli L, Peverieri GS, Paoli F, Bosio G, Venanzio D, Giacometto E, Roversi PF (2019) A new device for auto-disseminating entomopathogenic fungi against Popillia japonica: a study case. Bull Insectol 72:219–225
Blumenfeld AJ, Eyer PA, Husseneder CMoJ, Johnson LNL, Wang C, Kenneth Grace J, Chouvenc T, Wang S, Vargo EL (2021) Bridgehead effect and multiple introductions shape the global invasion history of a termite. Commun Biol. https://doi.org/10.1038/s42003-021-01725-x
Bouckaert R (2016) Phylogeography by diffusion on a sphere: whole world phylogeography. PeerJ 4:e2406. https://doi.org/10.7717/peerj.2406
Bouckaert RR, Drummond AJ (2017) bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol Biol 17:42. https://doi.org/10.1186/s12862-017-0890-6
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537. https://doi.org/10.1371/journal.pcbi.1003537
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinform 10:421. https://doi.org/10.1186/1471-2105-10-421
CERIS Pest Tracker: Exotic Pest Reporting (2020) Maps of Japanese beetle, Popillia japonica—Pest tracker—CAPS services. https://pest.ceris.purdue.edu/map.php?code=INBPAZA
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chapuis MP, Lecoq M, Michalakis Y, Loiseau A, Sword GA, Piry S, Estoup A (2008) Do outbreaks affect genetic population structure? a worldwide survey in Locusta migratoria, A pest plagued by microsatellite null alleles. Mol Ecol 17:3640–3653. https://doi.org/10.1111/j.1365-294X.2008.03869.x
CFIA (2009) Questions and answers: Japanese beetle (Popillia japonica). http://www.inspection.gc.ca/english/plaveg/pestrava/popjap/queste.shtml. Accessed 4 November 2021
CFIA (2017) Popillia Japonica (Japanese beetle)—fact sheet. URL https://inspection.canada.ca/plant-health/invasive-species/insects/japanese-beetle/fact-sheet/eng/1328165101975/1328165185309 Accessed 11 November 2022
CFIA (2020) Appendix 1: regulatory status of areas in Canada and the United States for Japanese beetle (Popillia japonica). http://www.inspection.gc.ca/plants/plant-pests-invasive-species/directives/horticulture/d-96-15/appendix-1/eng/1346826626609/1346826990603.
Clair DJ, Kramer VL (1989) Japanese beetle. In: Dahlsten DL, Garcia R (eds) Eradication of exotic pests: analysis with case histories. Yale University Press, New Haven, pp 89–107
Clement M, Snell Q, Walker P, Posada D, Crandall K (2002) TCS: estimating gene genealogies. In: Parallel and distributed processing symposium, international. IEEE Comput Soc, pp 0184–0184
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Davis JJ (1920) The green Japanese beetle problem. J Econ Entomol 13(2):185–194. https://doi.org/10.1093/jee/13.2.185
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J Royal Statistical Society Series B 39:1–38. https://www.jstor.org/stable/2984875
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
EFSA, Schrader G, Camilleri M, Ciubotaru RM, Diakaki M, Vos S (2019) Pest survey card on Popillia japonica. EFSA Support Publ 16:1568E. https://doi.org/10.2903/sp.efsa.2019.EN-1568
EPPO (2012) Situation of Diabrotica virgifera virgifera in the EPPO region. https://www.eppo.int/ACTIVITIES/plant_quarantine/shortnotes_qps/diabrotica_virgifera
EPPO (2014) First report of Popillia japonica in Italy. EPPO reporting service 2014/179. https://gd.eppo.int/reporting/article-3272
EPPO (2016) PM 9/21(1) Popillia japonica: procedures for official control. EPPO Bull 46:543–555. https://doi.org/10.1111/epp.12345
EPPO (2017) First report of Popillia japonica in Switzerland. EPPO reporting service Num. article: 09–160
EPPO (2018) Popillia japonica found in Vancouver (British Columbia, Canada). EPPO reporting service Num. article: 05–092
EPPO (2020) EPPO Datasheet: Popillia japonica. https://gd.eppo.int/taxon/POPIJA/datasheet
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130. https://doi.org/10.1111/j.1365-294X.2010.04773.x
EU Commission Delegated Regulation (2019) 1702 of 1 August 2019 supplementing regulation (EU) 2016/2031 of the European Parliament and of the Council by establishing the list of priority pests. Official journal of the European Union L. 260/8, 11/10/2019
EU Convention of Biological Diversity (2002) Relevant international instruments—National invasive alien species strategies and action plans. https://www.cbd.int/decisions/cop/6/23/14.a
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Facon B, Genton BJ, Shykoff J, Jarne P, Estoup A, David P (2006) A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol Evol 21:130–135. https://doi.org/10.1016/j.tree.2005.10.012
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. https://doi.org/10.1093/genetics/164.4.1567
Fleming WE (1972) Biology of the Japanese beetle. Technical bulletin, vol 1449. US Department of Agriculture, Washington
Fleming WE (1976) Integrating control of the Japanese beetle: a historical review, vol 1545. US Department of Agriculture, Agricultural Research Service
Floerl O, Inglis GJ, Dey K, Smith A (2009) The importance of transport hubs in stepping-stone invasions. J Appl Ecol 46:37–45. https://doi.org/10.1111/j.1365-2664.2008.01540.x
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial Cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Fox H (1932) The distribution of the Japanese beetle in 1930 and 1931, with special reference to the area of continuous infestation. J Econ Entomol 25:396–407. https://doi.org/10.1093/jee/25.2.396
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth hitchhiking and background selection. Genetics 147:915–925. https://doi.org/10.1093/genetics/147.2.915
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372. https://doi.org/10.2307/2532296
Harry M, Solignac M, Lachaise D (1998) Molecular evidence for parallel evolution of adaptive syndromes in Fig-BreedingLissocephala (Drosophilidae). Mol Phylogenet Evol 9:542–551. https://doi.org/10.1006/mpev.1998.0508
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Hungate BA, Kearns DN, Ogle K, Caron M, Marks JC, Rogg HW (2016) Hydrogen isotopes as a sentinel of biological invasion by the Japanese beetle, Popillia japonica (Newman). PLoS ONE 11:e0149599. https://doi.org/10.1371/journal.pone.0149599
Kalinowski SM, Taper S (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7:991–995. https://doi.org/10.1007/s10592-006-9134-9
Klein M (2008) Popillia japonica (Japanese beetle) Invasive Species Compendium. CAB International, Wallingford. https://doi.org/10.1079/ISC.43599.20203373917
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Liebhold AM, Work TT, McCullough DG, Cavey JF (2006) Airline baggage as a pathway for alien insect species invading the United States. Am Entomol 52:48–54. https://doi.org/10.1093/ae/52.1.48
Lombaert E, Guillemaud T, Cornuet JM, Malausa T, Facon B et al (2010) Bridgehead effect in the worldwide invasion of the biocontrol harlequin ladybird. PLoS ONE 5(3):e9743. https://doi.org/10.1371/journal.pone.0009743
Marianelli L et al (2018) Entomopathogenic nematodes as potential biological control agents of Popillia japonica (Coleoptera, Scarabaeidae) in Piedmont Region (Italy). J Appl Entomol 142:311–318. https://doi.org/10.1111/jen.12470
Marianelli L et al (2019) Long-lasting insecticide-treated nets: a new integrated pest management approach for Popillia japonica (Coleoptera: Scarabaeidae). Integr Environ Assess Manag 15:259–265. https://doi.org/10.1002/ieam.4107
Martins A, Simões N (1985) Population Dynamics of the Japanese beetle (Coleoptera, Scarabaeidae) in Terceira Island – Azores. Arquipélago 6:57–62. https://repositorio.uac.pt/bitstream/10400.3/998/1/Population%20dynamics%20of%20the%20japanese%20beetle%20%28Coleoptera%20-%20Scarabaeidae%29%20in%20Terceira%20island%20-%20Azores.pdf
Mazza G et al (2017) Hexamermis popilliae n. sp. (Nematoda: Mermithidae) parasitizing the Japanese beetle Popillia japonica Newman (Coleoptera: Scarabaeidae) in Italy. Syst Parasitol 94:915–926. https://doi.org/10.1007/s11230-017-9746-0
Mori N, Santoiemma G, Glazer I, Gilioli G, Ciampitti M, Cavagna B, Battisti A (2022) Management of Popillia japonica in container-grown nursery stock in Italy. Phytoparasitica 50:83–89. https://doi.org/10.1007/s12600-021-00948-2
Nardi F, Carapelli A, Dallai R, Roderick GK, Frati F (2005) Population structure and colonization history of the olive fly, Bactrocera oleae (Diptera, Tephritidae). Mol Ecol 14:2729–2738. https://doi.org/10.1111/j.1365-294X.2005.02610.x
Orlov YL, Potapov VN (2004) Complexity: an internet resource for analysis of DNA sequence complexity. Nucleic Acids Res 32:W628–W633. https://doi.org/10.1093/nar/gkh466
Paoli F et al (2017) Differential susceptibility of Popillia japonica 3rd instars to Heterorhabditis bacteriophora (Italian strain) at three different seasons. Biocontrol Sci Technol 27:439–444. https://doi.org/10.1080/09583157.2017.1285866
Pavesi M (2014) Popillia japonica specie aliena invasiva segnalata in Lombardia. L’informatore Agrario 32:53–55
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Potter DA, Held DW (2002) Biology and management of the Japanese beetle. Annu Rev Entomol 47:175–205. https://doi.org/10.1146/annurev.ento.47.091201.145153
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Ramasamy RK, Ramasamy S, Bindroo BB, Naik VG (2014) STRUCTURE PLOT: a program for drawing elegant STRUCTURE bar plots in user friendly interface. Springerplus 3:431. https://doi.org/10.1186/2193-1801-3-431
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283. https://doi.org/10.1111/j.1558-5646.1995.tb04456.x
Raymond M, Rousset F (2001) GENEPOP (version 3.3): population genetics software for exact tests and ecumenicism. Updated from Raymond, M., and F. Rousset. 1995.
Regione Piemonte (2021) D.D. n. 872 del 12 Ottobre 2021, "Aggiornamento dell'area delimitata per la presenza di Popillia japonica Newman in Piemonte" http://www.regione.piemonte.it/governo/bollettino/abbonati/2021/41/attach/dda1700000872_10400.pdf.
Roderick GK, Navajas M (2003) Genes in new environments: genetics and evolution in biological control. Nat Rev Genet 4:889–899. https://doi.org/10.1038/nrg1201
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Saito O (2009) Environmental and economic scenario analysis of the redundant golf courses in Japan. Int J Humanit Soc Sci 3:1865–1871
Schierenbeck KA, Aïnouche ML (2006) The role of evolutionary genetics in studies of plant invasions. In: Cadotte MW, McMahon SM, Fukami T (eds) Conceptual ecology and invasion biology: reciprocal approaches to nature. Springer, Dordrecht, pp 193–221. https://doi.org/10.1007/1-4020-4925-0_9
Scott MJ et al (2020) Genomic analyses of a livestock pest, the New World screwworm, find potential targets for genetic control programs. Commun Biol 3:424. https://doi.org/10.1038/s42003-020-01152-4
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701. https://doi.org/10.1093/aesa/87.6.651
Sipolski SJ, Datson SW, Reding M, Oliver JB, Alm SR (2019) Minimizing bee (Hymenoptera: Apoidea) bycatch in Japanese beetle traps. Environ Entomol 48(5):1203–1213. https://doi.org/10.1093/ee/nvz098
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. https://doi.org/10.1093/genetics/123.3.585
Torrini G et al (2020) Evaluation of indigenous entomopathogenic nematodes as potential biocontrol agents against Popillia japonica (Coleoptera: Scarabaeidae) in Northern Italy. InSects 11:804. https://doi.org/10.3390/insects11110804
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115–e115. https://doi.org/10.1093/nar/gks596
USDA: Animal and Plant Health Inspection Service (2020) Japanese beetle distribution in the U.S. https://www.aphis.usda.gov/aphis/ourfocus/planthealth/plant-pest-and-disease-programs/pests-and-diseases/japanese-beetle/japanese-beetle.
USDA: Animal and Plant Health Inspection Service (2022) Japanese beetle. URL: https://www.aphis.usda.gov/aphis/ourfocus/planthealth/plant-pest-and-disease-programs/pests-and-diseases/japanese-beetle/japanese-beetle. Last access: 11/11/2022
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform 13:134. https://doi.org/10.1186/1471-2105-13-134
Acknowledgements
The authors would like to thank all collectors for their valuable contributions: Katsura Ito, Isao Takenaka, Shin-ya Ohba, Haruki Tatsuta, Ken Funayama, Yoshihiro Kunitomo, Kouichi Oyamada, Ryusuke Ishikawa, Kiichi Shimizu, Shingo Tanaka, Takuma Takanashi, Satoshi Kitabayashi, Koji Noge, Yoriko Saeki, Graham S. Thurston, Allison Barr, Ronald Neville, Jody, Green, Alan Eaton, Bill Jones, Claudio Gratton, Diana N. Kearns, David Held, Johnson Donald Steinkraus, Frank Krell, Sarah Zukoff, Paulo Castrovillo, Albrecht Koppenhöfer, R. C. McDonald, Charlene Donahue, Doug Terhune, Nadeer N. Youssef, Paulo Borges, Cristina Marazzi, Chun Lin Li and Chen Ri-Zhao. The authors would also thank Ilaria Cutino and Chiara Leo for their support.
Funding
This work has received funding from the regional project “COBIPO—Biological Control of Popillia japonica” [Piedmont Region (D n. 910 – 2015/12/29)] and European Union’s Horizon 2020 research and innovation programme “IPM POPILLIA” Integrated pest Management of the invasive Japanese Beetle, Popillia japonica, under grant agreement No. 861852.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This study does not contain any experiments using any animal species that require ethical approval.
Consent to participate
Consent was given by all participants included in the study.
Consent for publication
All authors consent to the publication of this manuscript, figures, tables and supporting materials in Journal of Pest Science.
Additional information
Communicated by Nicolas Desneux.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10340_2023_1653_MOESM1_ESM.xlsx
Supplementary Table 1 (Spreadsheet “Samples”) List of all samples used in this work with sampling dates and sites, geographical coordinates, mitochondrial haplotype found and sample groups assigned for microsatellite analysis. (Spreadsheet “Primers”) Primers developed in this work and annealing temperature (Tm) used. Microsatellite primer pairs were annotated with parameters of their reference allele.1 (XLSX 459 KB)
10340_2023_1653_MOESM2_ESM.tif
Supplementary Fig 2 Null allele estimation (A) Hardy-Weinberg test for heterozygote deficiency. (B) Fst per population and per locus considering the null allele (C) Genetic distance between populations considering null allele. (TIF 751 KB)
10340_2023_1653_MOESM3_ESM.tif
Microsatellite analysis of Popillia japonica from different area subsets. A All areas, 2 clusters (redrawn from Fig.1A); B all areas, 4 clusters (redrawn from Fig.1B); C: Japan only, 2 clusters; D: Japan and U.S.A. only, 2 clusters; E: U.S.A., Azores and Italy only, 2 clusters; F: U.S.A., Azores and Italy only, 3 clusters. Locations/individuals are vertically aligned across panels. (TIF 445 KB)
10340_2023_1653_MOESM4_ESM.tif
Single tree from COX I obtained in Bayesian and maximum likelihood analyses. Posterior probabilities and bootstrap values are labeled above the appropriate internodes. (TIF 12851 KB)
10340_2023_1653_MOESM5_ESM.tif
Single tree from CytB obtained in Bayesian and maximum likelihood analyses. Posterior probabilities and bootstrap values are labeled above the appropriate internodes. (TIF 11440 KB)
10340_2023_1653_MOESM6_ESM.tif
Phylogeographical analysis (A) single tree from the combined analysis of COX I, CytB and geographical coordinates that was obtained in Bayesian and maximum likelihood analyses. Posterior probabilities and bootstrap values are labeled above the appropriate internodes. The color code was the same28as that adopted in Figure 4. (TIF 413 KB)
10340_2023_1653_MOESM7_ESM.tif
Phylogeographical analysis (B) Maps showing the geographical location of sample groups in Japan and North America. (TIF 2024 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Strangi, A., Paoli, F., Nardi, F. et al. Tracing the dispersal route of the invasive Japanese beetle Popillia japonica. J Pest Sci 97, 613–629 (2024). https://doi.org/10.1007/s10340-023-01653-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-023-01653-1