
Abstract
The application of temperature gradient interaction chromatography (TGIC) as an advanced technique for the characterisation of polymers is discussed, in comparison to other liquid chromatography techniques and in particular the ubiquitous size exclusion chromatography. Specifically, the use of reversed-phase TGIC for the interrogation of complex branched polymers and normal-phase TGIC for characterisation of high-molar mass end-functionalised polymers is highlighted.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction to Interaction Chromatography of Polymers
Temperature Gradient Interaction Chromatography (TGIC) is an Interaction Chromatography (IC) technique for the characterisation of polymers in which separation is dominated by enthalpic interactions between the analyte and stationary phase. Other chromatographic techniques used for the characterisation of polymers include gradient polymer elution chromatography (GPEC), barrier and size exclusion chromatography (SEC) gradient methods and liquid chromatography at the critical conditions (LCCC) in traditional precipitation–redissolution mode. Details of these techniques have been reviewed elsewhere [1,2,3].
The earliest examples of IC characterisation of polymers were performed in the late 1960s by thin-layer chromatography [4, 5] and 10 years later by column liquid chromatography [6, 7]. In these early attempts, the successful separation of homopolymers of differing molar mass and copolymers with varying chemical composition was achieved. IC methodologies are a useful complement to the almost ubiquitous size exclusion chromatography (SEC) and may overcome the intrinsic limitation of the entropy-controlled methods, whereby (in SEC) separation is dependent upon polymer size in solution, i.e. the solvodynamic volume in a given solvent and temperature [8,9,10]. Other methods for size-based separation are hydrodynamic chromatography (HDC) [11] and flow field-flow fractionation (FlFFF) [12]. In SEC the polymers, dissolved in a good solvent should not interact with/adsorb to the column packing materials, and the distribution equilibrium is solely regulated by the entropic exclusion of polymer chains from pores in the packing beads, according to their size. In SEC mode, smaller chains will elute later due to longer retention in the pores, while larger chains elute earlier and all analytes are eluted before the injection solvent peak [13]. The most significant limitation of SEC is that polymers with the same hydrodynamic volume can sometimes have a different molar mass, due to differences in molecular architecture or different chemical composition. In such cases, SEC is incapable of separating polymers with similar hydrodynamic volume but different chemical or architectural characteristics.
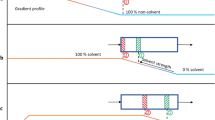
In contrast to SEC, IC exploits the enthalpic interaction of analytes with the surface of the column packing materials, and the equilibrium of the analytes between the mobile and the stationary phases is dependent not only on molar mass but also on the chemical and structural composition of the polymers. The eluents in IC are commonly weakly polar, to promote an appropriate interaction with the stationary phase [13]. In IC elution mode the polymers will elute after the injection solvent peak, and the interaction energy with the stationary phase is expected to be proportional to molar mass [14]. Therefore, the retention of a polymer increases exponentially with molar mass. Under specific conditions (solvent strength and temperature) the so-called critical adsorption point (CAP) for a particular homopolymer can be found [14] where the entropic (exclusion—SEC mode) and enthalpic (interaction—IC mode) effects cancel each other out and all samples of the homopolymer will elute in correspondence to the total void volume, irrespective of molar mass. The LC method utilizing this phenomenon is called liquid chromatography at the critical condition (LCCC) [1]. The three possible elution modes are illustrated in Fig. 1. It is possible to switch between IC and SEC separation modes, passing through the CAP, with the same stationary phase by changing either the solvent strength/polarity or column temperature. In TGIC, the optimal composition is fixed near the CAP condition, and the column temperature is tuned to change the separation mode from IC to SEC [13]. In a typical TGIC experiment, the temperature is raised to gradually increase polymer desorption and promote elution, given that adsorption of polymer is generally an exothermic process [1]. However, it should be noted that in a few rare cases polymer–eluent–column systems have been reported with an inverse temperature dependence [15,16,17].

Polymer molar mass (M) versus retention volume in three chromatographic separation modes: size exclusions chromatography (SEC), liquid chromatography at critical condition (LCCC) and interaction chromatography (IC). Reprinted from ref [13] with permission from Wiley
TGIC and Its Applications
The earliest reports of TGIC by Chang et al. describe the successful and enhanced (compared to SEC) separation of linear homopolymers according to their molar mass [18,19,20,21,22]. However, the real potential of TGIC is in exploiting the enthalpic nature of the interaction between polymer samples and the stationary phase, to enable separation according to architecture (chain branching), tacticity, chemical composition and functionalisation. In common with many other IC techniques, TGIC can be carried out under reversed-phase (RP) and normal-phase (NP) conditions.
Reversed-Phase TGIC
In reversed-phase (RP) conditions, the stationary phase is non-polar, e.g. C18 modified silica, while the mobile phase is more polar than the stationary phase. RP-TGIC resolves polymer samples based on molar mass instead of hydrodynamic volume, which is particularly useful for the analysis of branched polymers where variation in structure may result in a significant difference in molar mass, but almost no difference in hydrodynamic volume. In such cases, SEC analysis is incapable of detecting dispersity in architecture or molar mass. In the past decade, the advent of RP-TGIC has been crucial in underpinning the development of models to describe the correlation of polymer architecture and polymer rheology. The presence of even small quantities of defects and by-products can have significant implications when relating the polymer structure to experimental rheology whereby discrepancies make the validation and subsequent modification of theoretical models impossible [23].
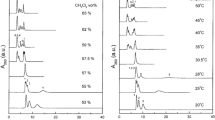
RP-TGIC has been used to investigate the structural dispersity for a variety of branched architectures including star-branched polymers [24,25,26,27,28], mikto-arm star block copolymers [25, 29], H-shaped polymers [30, 31], comb-branched polymers [32, 33], dendritically branched polymers [34] and hyperbranched polymers [35]. The power of this technique is illustrated in Fig. 2 which shows the SEC (Fig. 2a) and RP-TGIC (Fig. 2b) analysis of a polybutadiene G1 DendriMac [36]. Such complex, hierarchically branched polymers have been critical in structure–property correlation studies. It is clear that SEC analysis is indicating a well-defined, narrow dispersity product—analysis gave a dispersity of 1.04. However, RP-TGIC analysis revealed the presence of a low molar mass species at lower retention volume (the order of elution is reversed in TGIC compared to SEC) which was subsequently shown to be the G1 DendriMac with one outer arm missing and this defect was found to comprise nearly 14% by mass of the product. RP-TGIC is much less universal than SEC in so much that each polymer analysed requires chromatographic conditions (eluent, temperature and temperature gradient) to be optimised, which can be time-consuming. However, as illustrated, RP-TGIC has proved to be crucial in the accurate characterisation of complex branched polymers.

Chromatograms of polybutadiene G1 DendriMac obtained by size exclusion chromatography (a) and reversed-phase temperature gradient interaction chromatography (b) [34]
Normal Phase (NP) TGIC
On the other hand exploits a polar stationary phase, commonly bare silica or diol bonded silica, and a less polar mobile phase. Until recently there were very few examples in the literature of the application of this technique, beyond demonstrating that NP-TGIC can also be used for the analysis of molar mass distributions. However, more recently, the significant potential of NP-TGIC for the analysis of end-functionalised polymers has been demonstrated and it is particularly well-suited for the analysis of end-functionalised polymers of high molar mass, for which the accuracy of more common techniques, such as MALDI and NMR, is inherently less reliable. It is worth noting that the presence of chain-end functionality, in most cases, has no impact on the hydrodynamic volume of a polymer chain, and therefore SEC is of no use in this context. Thus, NP-TGIC has been used for both the identification and the quantification of chain-end functionality. This was first demonstrated in 2001 by Chang et al. who used NP-TGIC for the analysis of hydroxyl chain-end-functionalised polystyrene homopolymers of two different molar masses (11 and 105 kg mol−1). Chang managed to successfully resolve in a single run the polystyrene samples, both in terms of molar mass and chain end functionality, separating the unfunctionalised chains from the functionalised ones [37] (Fig. 3).

NP-TGIC separation of polystyrene samples with different end groups (hydrogen terminated vs hydroxyl terminated). Reprinted from ref [37] with permission from Elsevier
The NP-TGIC separation of homopolymers with different functional groups has been successfully achieved by our group [38, 39], showing that it is possible to follow chain end functional group transformations and to obtain successful separation of chains with a molar mass of up to 200,000 g mol−1 which differ only by the presence of a single primary alcohol functionality at the chain-end [39]. At such molar masses, any attempt to identify the presence of, or to attempt to quantify the extent of end-functionalization by NMR or MALDI-ToF MS would be an act of futility.
Additional Applications of TGIC
Although (in the opinion of these authors) the greatest impact of TGIC has been in the characterisation of complex branched polymers by RP-TGIC and the analysis of high-molar mass end-functionalised polymers by NP-TGIC, TGIC has been used for the characterisation of polymers in a number of other specific cases. Thus, TGIC has been used to characterise a variety of block copolymers [40,41,42] and for the separation of polymers according to tacticity, where samples of isotactic, heterotactic and syndiotactic poly(ethyl methacrylate) of similar molar mass were resolved according to stereoregularity [43]. Moreover, in some cases, the characterisation and resolution of complex branched polymers has been enhanced by taking advantage of an H/D isotope effect. Thus, it has been shown that reversed-phase interaction chromatography is sensitive to this isotope effect and deuterated polymers are less strongly retained than their non-deuterated analogue. This phenomenon has been exploited in the RP-TGIC characterisation of H-shaped polymers with a deuterated cross-bar and hydrogenous arms [30] and in a 2D LC experiment (NP-TGIC/RP-LC) for the characterisation of comb-branched polymers with a hydrogenous backbone and deuterated arms [33]. Finally, Cong and co-workers developed a high-temperature TGIC (HT-TGIC) methodology enabling the high-resolution separation of poly(ethylene–octene) copolymers according to their comonomer distribution and chemical composition [44].
Limitations and Opportunities
TGIC offers significant advantages over SEC in offering the possibility to separate polymers on the basis of molar mass (RP-TGIC) and/or chemical composition/functionality. Thus, TGIC has been shown to be a particularly powerful technique for the characterisation of complex branched polymers, in identifying the presence of and quantifying the amount of defect structures. NP-TGIC is capable of characterising high-molar mass end-functionalised polymers, in some cases resolving polymers of identical molar mass on the basis of a single functional group, even in polymers with a molar mass of up to 200,000 g mol−1, when alternative techniques such as NMR and MALDI-ToF MS offer no insight. It has been also shown that TGIC gives lower band-broadening compared to SEC, when used for molar mass separation, thus providing for higher resolution [21]. In contrast to other advanced liquid chromatography methods, the isocratic elution in TGIC limits the background signal drift encountered in solvent gradient elution methods, enabling the use of differential detection methods (e.g. UV, light scattering and viscometry), with appropriate temperature control at the detectors.
However, TGIC is not without its limitations. The application of a temperature gradient can lead to significant baseline drift in a differential refraction index detector—such detectors being extremely sensitive to changes in temperature. Moreover, a temperature gradient cannot vary the solvent strength of the eluent as much as a variation in solvent composition, for which infinite combinations are possible, either using binary or ternary solvent mixtures. Therefore TGIC is suitable for the analysis of polymer systems that need a fine control instead of a large change of the solvent strength [1]. Furthermore, possibly the most limiting characteristic of TGIC, and the one that has inhibited the wide uptake of TGIC, is the fact that it is not a ‘universal method’ like SEC, where many different polymers can be characterised using a common solvent such as THF. In the case of TGIC, it is necessary to tune the chromatographic conditions (solvent, flow rate, temperature, temperature gradient and in some cases the nature of the column) for each type of polymer. The development of suitable conditions for a polymer or copolymer which has not previously been subjected to TGIC can be a very time-consuming process.
Despite these limitations, RP-TGIC has played a fundamental role in understanding the impact of chain-branching on the rheology of polymers, with significant implications for industrial-scale processing of commodity polymers, and NP-TGIC has the potential to be of huge value in the characterisation of end-functionalised polybutadiene and solution styrene–butadiene rubbers which are increasingly used in tyre rubber formulations. Such polymers are manufactured on a huge scale and frequently have a molar mass > 300,000 g mol−1. Attempts to characterise the extent of end-capping in such cases, by other techniques, are practically impossible.
References
Chang T (2018) Chromatographic separation of polymers. Recent progress in separation of macromolecules and particulates, vol 1281. ACS Symposium Series, American Chemical Society, Washington, pp 1–17
Radke W (2014) Polymer separations by liquid interaction chromatography: principles—prospects—limitations. J Chromatogr A 1335:62–79. https://doi.org/10.1016/j.chroma.2013.12.010
Striegel AM (2020) Method development in interaction polymer chromatography. TrAC, Trends Anal Chem 130:115990. https://doi.org/10.1016/j.trac.2020.115990
Inagaki H, Matsuda H, Kamiyama F (1968) Determination of compositional heterogeneity in copolymers by thin layer chromatography. I. Preliminary results for styrene-acrylate copolymers. Macromolecules 1(6):520–525. https://doi.org/10.1021/ma60006a013
Belenkii BG, Gankina ES (1970) Thin-layer chromatography of polymers introductory lecture. J Chromatogr A 53(1):3–25. https://doi.org/10.1016/S0021-9673(00)86699-6
Teramachi S, Hasegawa A, Shima Y, Akatsuka M, Nakajima M (1979) Separation of styrene-methyl acrylate copolymer according to chemical composition, using high-speed liquid chromatography. Macromolecules 12(5):992–996. https://doi.org/10.1021/ma60071a042
Armstrong DW, Bui KH (1982) Nonaqueous reversed-phase liquid chromatographic fractionation of polystyrene. Anal Chem 54(4):706–708. https://doi.org/10.1021/ac00241a024
Striegel A, Yau W, Kirkland J, Bly D (2009) Modern size-exclusion liquid chromatography, 2nd edn. Wiley, Hoboken, New Jersey
Striegel AM (2017) Chapter 10—size-exclusion chromatography. In: Fanali S, Haddad PR, Poole CF, Riekkola M-L (eds) Liquid chromatography, 2nd edn. Elsevier, Amsterdam, pp 245–273
Berek D (2010) Size exclusion chromatography—a blessing and a curse of science and technology of synthetic polymers. J Sep Sci 33(3):315–335. https://doi.org/10.1002/jssc.200900709
Striegel AM, Brewer AK (2012) Hydrodynamic chromatography. Annu Rev Anal Chem 5(1):15–34. https://doi.org/10.1146/annurev-anchem-062011-143107
Podzimek S (2011) Light scattering, size exclusion chromatography and asymmetric flow field flow fractionation: powerful tools for the characterization of polymers, proteins and nanoparticles. Wiley, Hoboken, New Jersey
Chang T (2005) Polymer characterization by interaction chromatography. J Polym Sci Part B: Polym Phys 43(13):1591–1607. https://doi.org/10.1002/polb.20440
Skvortsov A, Trathnigg B (2003) Martin’s rule revisited: its molecular sense and limitations. J Chromatogr A 1015(1):31–42. https://doi.org/10.1016/S0021-9673(03)01207-X
Lochmüller CH, Moebus MA, Liu Q, Jiang C, Elomaa M (1996) Temperature effect on retention and separation of poly(ethylene glycol)s in reversed-phase liquid chromatography. J Chromatogr Sci 34(2):69–76. https://doi.org/10.1093/chromsci/34.2.69
Teramachi S, Matsumoto H, Kawai T (2005) Direction of temperature gradient for normal-phase temperature gradient interaction chromatography in polystyrene fractionation. J Chromatogr A 1100(1):40–44. https://doi.org/10.1016/j.chroma.2005.09.023
Matsumoto H, Kawai T, Teramachi S (2007) Direction of temperature gradient and retention mechanism in normal-phase temperature gradient interaction chromatography for poly(methyl methacrylate) fractionation. Polym J 39(5):458–463. https://doi.org/10.1295/polymj.PJ2006181
Lee HC, Chang T (1996) Polymer molecular weight characterization by temperature gradient high performance liquid chromatography. Polymer 37(25):5747–5749. https://doi.org/10.1016/S0032-3861(96)00510-1
Lee W, Lee HC, Chang T, Kim SB (1998) Characterization of poly(methyl methacrylate) by temperature gradient interaction chromatography with on-line light scattering detection. Macromolecules 31(2):344–348. https://doi.org/10.1021/ma9707805
Chang T, Lee HC, Lee W, Park S, Ko C (1999) Polymer characterization by temperature gradient interaction chromatography. Macromol Chem Phys 200(10):2188–2204. https://doi.org/10.1002/(SICI)1521-3935(19991001)200:10%3c2188::AID-MACP2188%3e3.0.CO;2-F
Lee W, Lee H, Cha J, Chang T, Hanley KJ, Lodge TP (2000) Molecular weight distribution of polystyrene made by anionic polymerization. Macromolecules 33(14):5111–5115. https://doi.org/10.1021/ma000011c
Lee W, Lee HC, Park T, Chang T, Chae KH (2000) Characterization of polyisoprene by temperature gradient interaction chromatography. Macromol Chem Phys 201(3):320–325. https://doi.org/10.1002/(SICI)1521-3935(20000201)201:3%3c320::AID-MACP320%3e3.0.CO;2-4
Hutchings LR (2012) Complex branched polymers for structure-property correlation studies: the case for temperature gradient interaction chromatography analysis. Macromolecules 45(14):5621–5639. https://doi.org/10.1021/ma3005422
Lee HC, Lee W, Chang T, Yoon JS, Frater DJ, Mays JW (1998) Linking reaction kinetics of star shaped polystyrene by temperature gradient interaction chromatography. Macromolecules 31(13):4114–4119. https://doi.org/10.1021/ma971751x
Park S, Cho D, Im K, Chang T, Uhrig D, Mays JW (2003) Utility of interaction chromatography for probing structural purity of model branched copolymers: 4-miktoarm star copolymer. Macromolecules 36(15):5834–5838. https://doi.org/10.1021/ma034603h
Agostini S, Hutchings LR (2013) Synthesis and temperature gradient interaction chromatography of model asymmetric star polymers by the “macromonomer” approach. Eur Polym J 49(9):2769–2784. https://doi.org/10.1016/j.eurpolymj.2013.06.021
Ahn S, Lee H, Lee S, Chang T (2012) Characterization of branched polymers by comprehensive two-dimensional liquid chromatography with triple detection. Macromolecules 45(8):3550–3556. https://doi.org/10.1021/ma2021985
Lee HC, Chang T, Harville S, Mays JW (1998) Characterization of linear and star polystyrene by temperature-gradient interaction chromatography with a light-scattering detector. Macromolecules 31(3):690–694. https://doi.org/10.1021/ma9710996
Cho D, Park S, Chang T, Avgeropoulos A, Hadjichristidis N (2003) Characterization of a 4-miktoarm star copolymer of the (PS-b-PI)3 PS type by temperature gradient interaction chromatography. Eur Polym J 39(11):2155–2160. https://doi.org/10.1016/S0014-3057(03)00154-X
Perny S, Allgaier J, Cho D, Lee W, Chang T (2001) Synthesis and structural analysis of an H-shaped polybutadiene. Macromolecules 34(16):5408–5415. https://doi.org/10.1021/ma001608v
Chen X, Rahman MS, Lee H, Mays J, Chang T, Larson R (2011) Combined synthesis, TGIC characterization, and rheological measurement and prediction of symmetric H polybutadienes and their blends with linear and star-shaped polybutadienes. Macromolecules 44(19):7799–7809. https://doi.org/10.1021/ma2011377
Chambon P, Fernyhough CM, Im K, Chang T, Das C, Embery J, McLeish TCB, Read DJ (2008) Synthesis, temperature gradient interaction chromatography, and rheology of entangled styrene comb polymers. Macromolecules 41(15):5869–5875. https://doi.org/10.1021/ma800599m
Ahn S, Im K, Chang T, Chambon P, Fernyhough CM (2011) 2D-LC characterization of comb-shaped polymers using isotope effect. Anal Chem 83(11):4237–4242. https://doi.org/10.1021/ac2005907
Hutchings LR, Kimani SM, Hoyle DM, Read DJ, Das C, McLeish TCB, Chang T, Lee H, Auhl D (2012) In silico molecular design, synthesis, characterization, and rheology of dendritically branched polymers: closing the design loop. ACS Macro Lett 1(3):404–408. https://doi.org/10.1021/mz300059k
Im K, Park S, Cho D, Chang T, Lee K, Choi N (2004) HPLC and MALDI-TOF MS analysis of highly branched polystyrene: resolution enhancement by branching. Anal Chem 76(9):2638–2642. https://doi.org/10.1021/ac035506p
Kimani SM, Hutchings LR (2008) A facile route to synthesize well-defined polybutadiene dendrimacs. Macromol Rapid Commun 29:633–637. https://doi.org/10.1002/marc.200700851
Lee W, Cho D, Chun BO, Chang T, Ree M (2001) Characterization of polystyrene and polyisoprene by normal-phase temperature gradient interaction chromatography. J Chromatogr A 910(1):51–60. https://doi.org/10.1016/S0021-9673(00)01163-8
Pagliarulo A, Hutchings LR (2018) End-functionalized chains via anionic polymerization: can the problems with using diphenylethylene derivatives be solved by using bisphenol F? Macromol Chem Phys 219(1):1700386. https://doi.org/10.1002/macp.201700386
Hutchings LR, Agostini S, Oti ME, Keth J (2015) Normal-phase (temperature gradient) interaction chromatography—a powerful tool for the characterisation of high molecular weight chain-end functionalised polymers. Eur Polym J 73:105–115. https://doi.org/10.1016/j.eurpolymj.2015.10.010
Park S, Park I, Chang T, Ryu CY (2004) Interaction-controlled HPLC for block copolymer analysis and separation. J Am Chem Soc 126(29):8906–8907. https://doi.org/10.1021/ja047385w
Park S, Cho D, Ryu J, Kwon K, Lee W, Chang T (2002) Fractionation of block copolymers prepared by anionic polymerization into fractions exhibiting three different morphologies. Macromolecules 35(15):5974–5979. https://doi.org/10.1021/ma0205313
Chung B, Park S, Chang T (2005) HPLC fractionation and surface micellization behavior of polystyrene-b-poly(methyl methacrylate). Macromolecules 38(14):6122–6127. https://doi.org/10.1021/ma050751r
Cho D, Park S, Chang T, Ute K, Fukuda I, Kitayama T (2002) Temperature gradient interaction chromatography and MALDI-TOF mass spectrometry analysis of stereoregular poly(ethyl methacrylate)s. Anal Chem 74(8):1928–1931. https://doi.org/10.1021/ac010995j
Cong R, deGroot W, Parrott A, Yau W, Hazlitt L, Brown R, Miller M, Zhou Z (2011) A new technique for characterizing comonomer distribution in polyolefins: high-temperature thermal gradient interaction chromatography (HT-TGIC). Macromolecules 44(8):3062–3072. https://doi.org/10.1021/ma200304e
Funding
There is no specific funding to report.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest to report.
Ethical approval
This article does not contain any studies with animals or human participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hutchings, L.R., Pagliarulo, A. Temperature Gradient Interaction Chromatography: A Perspective. Chromatographia 84, 813–818 (2021). https://doi.org/10.1007/s10337-021-04068-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-021-04068-1