
Abstract
A sensitive and reliable method using liquid chromatography–negative electrospray ionization mass spectrometry was developed for the simultaneous determination of chloramphenicol, florfenicol, and thiamphenicol at trace levels in animal feed. The analytes were extracted from grinded feed with ethyl acetate. Further the ethyl acetate was evaporated, residue resuspended in Milli-Q water, defatted with n-hexane, and solid phase extracted using BondELUT C18 cartridges. Separation was carried out on a C6 phenyl column with a mobile phase consisting of 0.1% formic acid in Milli-Q water and acetonitrile. The detector response was linear over the tested concentration range from 100 to 1000 µg kg−1. The recovery values for all analytes in feed were higher than 79% with RSD for repeatability and reproducibility in the ranges of 4.5–10.9% and 8.4–13.5%, respectively. CCα and CCβ varied between 76.8 and 86.1 µg kg−1, and between 111.3 and 159.9 µg kg−1, respectively. The results showed that this method is effective for the quantification of phenicols in non-target feed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antibiotics are widely used for therapeutic and prophylactic purposes in human and veterinary medicine and also to promote growth and increase feed efficiencies in food producing animals [1]. However, abused use of antibiotics and their presence in the food of animal origin are of concern due to development of resistance in target pathogens against antibiotics, induced allergic reactions in some hypersensitive individuals, and potential compromise of the human intestinal and immune systems [2, 3]. There is a diverse range of chemical substances with antimicrobial activity. Among them, phenicols, including chloramphenicol (CAP), thiamphenicol (TAP) and florfenicol (FF) are readily available broad-spectrum antibiotics. They are effective against a wide variety of Gram-negative and Gram-positive bacteria, including most anaerobic organisms. Chloramphenicol was first isolated in 1947 from cultures of Streptomyces venezulae, and synthetic production began in 1949 [4]. CAP was used widely for many years in veterinary practice, both therapeutically and prophylactically, due to its excellent pharmacokinetic and antibacterial properties. However, CAP is hematotoxic to humans and is associated with side effects such as bone marrow aplasia (loss of ability to produce blood cells), and therefore, aplastic anaemia. These effects are unrelated to dosage and generally fatal [5,6,7]. TAP and FF have been used as alternatives to CAP, since they do not have the same side effects. However, even though it is prohibited, CAP is still used because of its efficacy and relatively low cost as well as the availability and prevention of some infectious diseases in mammals, birds, bees and aquaculture. In addition to its illegal use, products of animal origin can contain CAP residues because of its occurrence in the environment [8]. According to the literature, chloramphenicol can still be found in several food matrices, suggesting its continued use [6, 9,10,11,12,13,14,15]. Besides, there is little information available regarding the occurrence of its analogues in foods of animal origin and environment. Therefore, sensitive and reliable methods for the analysis of phenicols are needed.
Various analytical methods have been reported for the determination of TAP and FF in food, such as gas chromatography (GC) [16, 17], liquid chromatography (LC) [18,19,20,21], GC–mass spectrometry (MS) [22, 23], LC–MS [24], and LC–MS/MS [25,26,27]. Sample preparation is critical to the validity of trace analysis. Previous investigations have set forth various types of pretreatment methods for fenicols in food before chromatographic determination, including liquid–liquid extraction (LLE) [28], solid-phase extraction (SPE) [29] or QuEChERS technique [27]. Conventional methods for extraction of organic analytes from food samples usually consist of a homogenisation step, followed by tedious liquid–liquid extraction procedures with one or more several clean-up steps and purification of the extract to remove co-extractants, before the sample is subjected to chromatographic separation. In the world of literature, there are many methods for analysing phenicols in food of animal origin, but methods for analysing phenicols in feed are still rare.
In the European Union, animal feeds must fulfil several rules laid down by current legislation [30,31,32]. The main rules give requirements for the composition, storage, transport, and usage of animal feeds. Cross-contamination can occur during production and handling in the feed mill, during transport, or on the farm. Carry-over of veterinary drugs during feed production may also cause the contamination of non-medicated feedstuffs. The use of antibiotics in feed for non-medicinal purposes was banned in the EU in 2006. According to the Regulation (EU) 2019/4 of the European Parliament and of the Council of 11 December 2018 on the manufacture, placing on the market and use of medicated feed, amending Regulation (EC) No 183/2005 of the European Parliament and of the Council and repealing Council Directive 90/167/EEC, by 28 January, 2023, the Commission shall indicate specific maximum levels for cross-contamination of non-target feed with active substances and methods for analysing active substances in feedingstuffs. Regarding the maximum levels of cross-contamination, delegated acts will be based on a scientific risk assessment carried out by the European Food Safety Authority [33]. Taking into account the European legislation for feed and carry-over problems during the production of medicated feeds, the objective of this article is to present a LC–MS method for the simultaneous identification and quantification of phenicols (florfenicol, tiamphenicol and chloramphenicol) in non-target feed.
Materials and Methods
Chemicals and Reagents
Florfenicol, thiamphenicol, chloramphenicol, and formic acid were from Sigma Aldrich (St. Louis, USA). HPLC-grade acetonitrile and methanol were purchased from Baker (Deventer, The Netherlands). Ethyl acetate was from Chempur (Piekary Śląskie, Poland) and n-hexane was from POCH (Gliwice, Poland). Purified water was prepared in-house with Milli-Q water system from Millipore (Bedford, MA, USA).
Two SPE cartridges BondElut C18 (500 mg, 3 mL) of Agilent (CA, USA) and Strata-X (polymeric sorbent-surface modified styrene divinylobenzene, 200 mg, 3 mL) from Phenomenex (Torrance, CA, USA) were used. An SPE manifold (J.T. Baker, Oklahoma, PA, USA) and a pump as a vacuum source were used.
Standard Solutions
Stock standard solutions of individual compound (1000 μg mL−1) were prepared by weighting of 20.0 mg of reference standard and dissolving in to 20 mL of acetonitrile. Working standard solutions (100 μg mL−1) were prepared by diluting the respective standard stock solutions in acetonitrile. Working standard solutions were prepared daily to avoid degradation problems.
Blank Samples
Samples were collected from feed mills and farms in Poland. Prior to use, the absence of veterinary drugs in all blank samples was confirmed by LC–MS analysis and by applying the developed method. Feed for different animal species (swine, poultry and cattle) have been collected, grinded and sieved through a sieve of 1 mm mesh size before analysis.
Calibration Curve Preparation
The test samples used for method development and validation were prepared by spiking the blank feed samples (5.00 g) with the working standard solutions (100 μg mL−1), and the target compounds at the concentration 100, 250, 500, 750 and 1000 µg kg−1 for matrix-matched calibration curves. The samples were shaken on a vortex mixer for 30 s, then allowed to stand at room temperature for 1 h to enable sufficient equilibration with the feed matrix.
Sample Preparation and Extraction
A sample feed of 5 ± 0.01 g was weighted into a 50 mL polypropylene centrifuge tubes. Next 20 mL of ethyl acetate was added and samples were shaken at room temperature for 25 min on a horizontal shaker and centrifuged at 3500×g for 15 min at 15 °C. Twelve millilitres of the supernatant was transferred to an amber glass conical tube and evaporated to dryness under nitrogen stream at 40 °C ± 5 °C. The residues were resuspended in 2 mL of mixture 30% methanol in Milli-Q water and shaken on a vortex mixer for 1 min, and next 5 mL of Milli-Q water was added. The resulting solution was transferred to a 15 mL centrifuge tube and shaken on a vortex mixer. Subsequently 2 mL of n-hexane was added to degrease the extract and was stirred manually for 1 min. Prepared samples were centrifuged at 3500×g 15 min, 15 °C. After centrifugation, the n-hexane layer was removed.
Clean-Up
For the clean-up step, the SPE apparatus and C18 BondELUT cartridges (500 mg, 3 mL) were used. Prior to the sample loading, the cartridges were preconditioned with 3 mL of methanol, and 3 mL of water. In total, 6 mL of sample solution prepared was loaded into the SPE cartridge. The cartridge was then washed with 3 mL of water and vacuum-dried for 5 min. The analytes were eluted with 3 mL of methanol. The eluate extract was evaporated to dryness under a nitrogen stream at 40 °C ± 5 °C. The residues were resuspended in 1 mL 30% methanol in Milli-Q water. Next, the extract was mixed on a vortex mixer, and transferred into autosampler vials for analysis.
LC–MS
For detection and quantification of phenicols, an Agilent LC–MS system consisting of an Agilent 1200 series liquid chromatograph and single quadrupole mass spectrometer Agilent 6140 (Agilent Technology, CA, USA) were used. The experiments were conducted in the negative electrospray mode with selected ion monitoring. The ChemStation software (Agilent Technology, CA, USA) controlled the LC–MS system and processed the data. The operating parameters were capillary voltage 2000 V, drying gas temperature (250 °C), drying gas flow (12 L min−1), and nebulising gas pressure (35 psi). Molecular masses of the precursor ions of FF, TAP and CAP were 321, 354, and 356 m/z, respectively. The separation of the phenicols was performed on a phenyl C6 (150 × 4.6 mm, 5 μm) column protected by a C6 guard column (4.0 × 3.0 mm, 5 μm), both from Phenomenex, operated at 20 °C. The mobile phase consisted of 0.1% formic acid in deionized water (solvent A) and acetonitrile (solvent B). The gradient used was 0–10 min, 20% B; 10–12 min, 36% B; 12–14 min, 36% B; before returning to 20% B in 2 min, with a final hold at 20% B until 20 min. The flow rate was 0.7 mL min−1 and the injection volume was 10 μL.
Validation Procedure
The proposed LC–MS–ESI method was in-house validation.The linearity of the method was evaluated using fortified blank feed samples. Good linearity was found by analysis of the feed samples spiked with standard solutions in the range 100–1000 µg kg−1 with the correlation coefficients higher than 0.998 for all analysed phenicols. The LOD, LOQ, CCα and CCβ parameters were estimated using the calibration curve procedure according to the ICH guideline and the European Commission Decision 2002/657/EC [34, 35]. The limit of detection (LOD) is the lowest concentration of analyte that the analytical process can reliably differentiate from background levels, while the limit of quantification (LOQ) is the lowest concentration of analyte that can be quantified. LOD and LOQ values were calculated from a signal-to-noise ratio (S/N) of 3 and 10, respectively. CCα was calculated by analysing 20 blank feed samples. Matrix-matched calibration curve was prepared and decision limit (CCα), and detection capability (CCβ) were determined according to the European Commission Decision 2002/657/EC for substances with no permitted limit. CCα was calculated with a statistical certainty of 1 − α (α = 1%) whereas CCβ was calculated with a statistical certainty of 1−β. CCβ was calculated as decision limit plus 1.64 times the corresponding standard deviation (β = 5%). Selectivity/specificity of the method was tested by analysing 20 blank feed samples to verify the absence of potential interfering endogenous compounds at the target analyte retention times. Intra-day precision was assessed by comparing the results of six replicates prepared on the same day at three different concentrations (100, 500 and 1000 µg kg−1). The procedure was repeated to determine inter-day precision by comparing results from samples prepared and analysed on three different days. Coefficients of variation (CV, %) and standard deviations (SD) were calculated for each level. Percentage recoveries were calculated as the measured content divided by the fortification level multiplied by 100. The measurement uncertainty was estimated using MUkit Measurement Uncertainty Kit 1.0.3.7 software (SYKE, Finnish Environmental Institute, Finlands). The matrix effect was checked by analysing feed samples for poultry and swine of 100 μg kg−1 concentration level and calculated by the equation proposed previously by Matuszewski et al. [36].
Results and discussion
Chromatographic Conditions
Chromatographic conditions were optimised to improve separation, sensitivity, and selectivity, taking into account the compounds investigated. Regarding the chromatographic analysis, due to the different physical–chemical characteristics of the phenicols selected, it was necessary to find a compromise to guarantee a sharp peak shape and reproducible retention times. The M–H ion was obtained as the precursor ion for individual fenicols antibiotics under the negative mode for both qualitative and quantitative determinations.
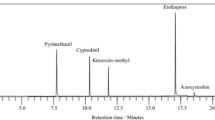
The mobile phase optimization was necessary to obtain satisfactory response for different compounds at particular concentration levels and for different feed matrices in both ionization polarities. For the analysis of CAP, TAP and FF form food and feed matrices, researchers usually use mobile phase formic acid, acetic acid, amonium acetate, amonium formate, amonium acetete with formic acid in water or pure water in combination with methanol or acetonitrile. The use of formic acid or acetic acid improved the response and peak shape of CAP, TAP and FF which are monitored in negative mode. Thus, we started the optimization procedure with an elution gradient of water and acetonitrile. With this scheme, it was possible to obtain satisfactory retention times for molecules in the negative mode. For the separation of phenicoles, researchers usually use modified C18 or C8 chromatographic columns such as LiChrospher, Symmetry Shield RP18, Zorbax Eclipse Plus, XTerra C18 and Hypersil C18-BD or Kinetex C8 [27, 37,38,39,40]. In this study, three chromatographic columns (Kinetex C18, Zorbax Eclipse C8 and phenyl C6) were investigated, and the best results (peak resolution and signal intensity) were obtained using the Phenomenex phenyl C6 column and mobile phase consisting of 0.1% formic acid in Milli-Q water and acetonitrile. The SIM and SCAN chromatograms of blank feed sample and feed sample spiked at 100 µg kg−1 for CAP, TAP and FF are shown in Figs. 1 and 2.

SIM chromatograms of blank feed sample

SIM chromatograms of feed sample spiked with thiamphenicol, florfenicol and chloramphenicol at a concentration of 100 μg kg−1
Sample Preparation
Animal feed is derived from a multitude of raw materials from plant and animal origin, as well as pharmeceutical and industrial sources. As feed ingredients are very dependent upon the animal species (poultry, swine, cattle, horse), developing a satisfactory procedure for the extraction and purification of amphenicoles from a feed matrix is one of the most time-consuming and complex process in optimising a method.
The extraction of analytes having different polar natures is always a difficult job. Various methods are found in the literature for simultaneous extraction of all the phenicols drugs in eggs, honey, milk, shrimps, fish, tissues, kidney and liver [40,41,42,43,44,45,46,47,48,49,50].
In the current study, various combinations of solvents, as described by previous reports, were evaluated. The method used by Xie et al. [51] was found to be the most relevant for the extraction of all phenicols. It consists of extraction with ethyl acetate/acetonitrile/ammonium hydroxide mixture, followed by hexane wash and without further clean-up step. However, using this method, CAP gave poor recoveries. A second extraction with mixture of acetonitrile/ethyl acetate/methanol was evaluated separately [52]. A third extraction used only ethyl acetate [2]. Ethyl acetate was better for the recovery of most of the remaining CAP and the rest of the pharmaceuticals as compared to previous two extraction methods. Ethyl acetate, due to its polarity, provides a high selective extraction of nonpolar compounds including fat [41]. After extraction feed samples were centrifuged,12 mL of extract was evaporated at the nitrogen stream. Residues were resuspended in 2 mL of 30% methanol in Milli-Q water, mixed, then added 5 mL of Milli-Q water and then 2 mL of n-hexane was added to defat the sample, then manually mixed and centrifuged.
SPE was used for sample clean-up. Two different SPE disposable cartridges were tested for clean-up and pre-concentration. The method of cartridges preparation was the same for C18 BondElut and Strata-X cartridges. The cartridges were conditioned with methanol and water, than samples were loaded. Cartridges were washed with water and dried under vacuum for 5 min. Next phenicoles were eluted with methanol. The experiment showed that better recoveries and pure extract was obtained with the BondELUT C18 cartridge than Strata-X cartridges. Therefore, for the purification of phenicols from feed matrix we selected BondELUT C18 cartridges.
Method Validation
The developed procedure was designed to obtain a method for detection and quantification of florfenicol, thiamphenicol and chloramphenicol in non-target feed. Due to the lack of a certified reference material for all analytes under investigation, the accuracy and precision of the method in terms of repeatability (RSD %, intra-day precision), reproducibility (RSD %, inter-day precision) and uncertainty were determined using blank feed samples fortified with known amounts of analytes. The accuracy and precision values were calculated at three validation levels for each compound on three different days. The mean extraction recoveries ranged from 80.7% to 93.1% for CAP, 79.3% to 101.4% for TAP, 83.2% to 93.9% for FF. Overall mean recoveries ranged from 86.8% to 92.7% for all the three analytes. The method precision was estimated in terms of RSD. The intra-day and inter-day variations calculated for all the analytes ranged from 4.5% to 10.9% and 8.4% to 13.5%, respectively. The values of CCα (decision limit) and CCβ (detection capability) were calculated from precision experiments for all the compounds and are presented in Table 1. The LOD and the LOQ values were estimated from blank feed extracts. Matrix effects were ± 36%, which is in compliance with SANTE/11945/2017 requirements [53]. The expanded uncertainty was estimated to be in the range of 29.8–34.8%, depending on the analyte. Specificity is the ability of the method to distinguish between the analyte of interest and the other substances (impurities or matrix components) that may be present in a test sample. In the evaluation, the specificity of blank feed samples was analysed by the MS detector. The results obtained with blank samples were compared with CAP, TAP and FF spiked samples and no interfering peaks were observed. The method was tested in in-house comparisons by analysing samples to which known concentrations of antibiotics were added by various analysts and by analysing real feed samples. The method was shown to be appropriate for all the analytes with acceptable accuracy and precision. All validation parameters are presented in Table 1.
Conclusions
A quantitative LC–MS–ESI method was optimized for the simultaneous determination of three fenicols (chloramphenicol, thiamphenicol and florfenicol) in feed samples. Good separation of all the compounds in less than 20 min and pure chromatograph image was obtained. The method has been successfully applied to the analysis of feed samples for poultry and swine. The results obtained using the method in the analysis of commercial feed samples showed that the method is able to quantify phenicols at carry-over level in different kinds of feeds.
References
Patyra E, Kwiatek K (2017) Determination of fluoroquinolones in animal feed by ion pair high-performance liquid chromatography with fluorescence detection. Ann Lett 50:1711–1720
Gavilán RE, Nebot C, Patyra E, Vazquez B, Miranda JM, Cepeda A (2019) Determination of florfenicol, thiamphenicol and cloramphenicol at trace levels in animal feed by HPLCMS/MS. Antibiotics 8(2):59
Jansen LJM, Bolck YJC, Berendsen BJA (2006) Feather segmatnation of discriminate between different enrofloxacin treatments in order to monitor off-label use in the poultry sector. Anal Bioanal Chem 408:495–502
Allen EH (1984) Review of chromatographic methods for chloramphenicol residues milk, eggs, and tissues from food-producing animals. J Assoc Off Anal Chem 68:990–999
Wongtavatchai J, McLean JG, Ramos F, Arnold D (2004) WHO food additives series: chloramphenicol, JECFA53. %3chttps://www.inchem.org/documents/jecfa/jecmono/v53je03.htm%3e. Accessed 14 May 2017
Samsonova JV, Cannavan A, Elliott CT (2012) A critical review of screening methods for the detection of chloramphenicol, thiamphenicol, and florfenicol residues in foodstuffs. Crit Rev Anal Chem 42:50–78
Guidi LR, Silva LHM, Fernandes C, Engeseth NJ, Gloria MB (2015) ALC-MS/MS determination of chloramphenicol in food of animal origin in Brazil. Sci Chromatogr 7:1–9
Guidi LR, Tette PAS, Fernandes C, Silva LHM, Glória BA (2017) Advances on the chromatographic determination of amphenicols in food. Talanta 162:324–338
Lu Y, Shen Q, Dai Z, Zhang H (2010) Multi-walled carbon nanotubes as solid-phase extraction adsorbent for the ultra-fast determination of chloramphenicol in egg, honey, and milk by fused-core C18-based high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 398:1819–1826
Martins-Junior HA, Bustillos OV, Pires MAF, Lebre DT, Wang AY (2006) Determination of chloramphenicol residues in industrialized milk and honey samples using LC–MS/MS. Quim Nova 29:586–592
Santos L, Barbosa J, Castilho MC, Ramos F, Ribeiro CAF, da Silveira MIN (2005) Determination of chloramphenicol residues in rainbow trouts by gas chromatography–mass spectometry and liquid chromatography–tandem mass spectometry. Anal Chim Acta 529:249–256
Sheridan R, Policastro B, Thomas S, Rice D (2008) Analysis and occurrence of 14 sulfonamide antibacterials and chloramphenicol in honey by solid-phase extraction followed by LC/MS/MS analysis. J Agric Food Chem 56:3509–3516
Verzegnassi L, Royer D, Mottier P, Stadler RH (2003) Analysis of chloramphenicol in honeys of different geographical origin by liquid chromatography coupled to electrospray ionization tandem mass spectrometry. Food Addit Contam 20:335–342
Wang H, Zhou XJ, Liu Y-Q, Yang H-M, Guo Q-L (2011) Simultaneous determination of chloramphenicol and aflatoxin M-1 residues in milk by triple quadrupole liquid chromatography-tandem mass spectrometry. J Agric Food Chem 59:3532–3538
Wu J, Chen L, Mao P, Lu Y, Wang H (2012) Determination of chloramphenicol in aquatic products by graphene-based SPE coupled with HPLC-MS/MS. J Sep Sci 35:3586–3592
Zhang SX, Sun FY, Li JC, Cheng LL, Shen JZ (2006) Simultaneous determination of florfenicol and florfenicol amine in fish, shrimp, and swine muscle by gas chromatography with a microcell electron capture detector. J AOAC Int 89:1437
Posyniak A, Zmudzki J, Niedzielska J (2003) Evaluation of sample preparation for control of chloramphenicol residues in porcine tissues by enzyme-linked immunosorbent assay and liquid chromatography. Anal Chim Acta 483:307–311
Giorgi M, Romani M, Bagliacca M, Mengozzi G (2000) Determination of thiamphenicol residues in albumin and yolk of hen eggs. J Vet Pharmacol Ther 23:397–399
Vue C, Schmidt LJ, Stehly GR, Gingerich WH (2003) Liquid chromatographic determination of florfenicol in the plasma of multiple species of fish. J Chromatogr B 780:111–117
Wrzesinski CL, Crouch LS, Endris R (2003) Determination of florfenicol amine in channel catfish muscle by liquid chromatography. J AOAC Int 86:515–518
Nagata T, Oka H (1996) Detection of residual chloramphenicol, florfenicol, and thiamphenicol in yellowtail fish muscles by capillary gas chromatography–mass spectrometry. J Agric Food Chem 44:1280–1286
Li P, Qiu YM, Cai HX, Kong Y, Tang YZ, Wang DN, Xie MX (2006) Simultaneous determination of chloramphenicol, thiamphenicol, and florfenicol residues in animal tissues by gas chromatography/mass spectrometry. J Chromatogr 24:14–18
van de Riet JM, Potter RA, Christie-Fougere M, Burns BG (2003) Simultaneous determination of residues of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in farmed aquatic species by liquid chromatography/mass spectrometry. J AOAC Int 86:510–514
Bogusz MJ, Hassan H, Al-Enazi E, Ibrahim Z, Al-Tufail M (2004) Rapid determination of chloramphenicol and its glucuronide in food products by liquid chromatography–electrospray negative ionization tandem mass spectrometry. J Chromatogr B 807:343–356
Forti AF, Campana G, Simonella A, Multari M, Scortichini G (2005) Determination of chloramphenicol in honey by liquid chromatography–tandem mass spectrometry. Anal Chim Acta 529:257–263
Zhang SX, Liu ZW, Guo X, Cheng LL, Wang ZH, Shen JZ (2008) Simultaneous determination and confirmation of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in chicken muscle by liquid chromatography–tandem mass spectrometry. J Chromatogr B 875:399–404
Śniegocki T, Sell B, Giergiel M, Posyniak A (2019) QuEChERS and HPLC–MS/MS combination for the determination of chloramphenicol in twenty two different matrices. Molecules. https://doi.org/10.3390/molecules24030384
Rřnning HT, Einarsen K, Asp TN (2006) Determination of chloramphenicol residues in meat, seafood, egg, honey, milk, plasma and urine with liquid chromatography–tandem mass spectrometry, and the validation of the method based on 2002/657/EC. J Chromatogr A 1118:226–233
Pfenning AP, Roybal JE, Rupp H, Turnipseed SB, Gonzales SA, Hurlbut JA (2000) Simultaneous determination of residues of chloramphenicol, florfenicol, florfenicol amine, and thiamphenicol in shrimp tissue by gas chromatography with electron capture detection. J AOAC Int 83:26–30
EC Directive 2002/32 (2002) Off J Eur Union L 140:10–21. https://eur-lex.europa.eu/legalcontent/EN/TXT/?uri=CELEX%3A32002L0032. Accessed 10 Dec 2018
EC Directive 2009/8 (2009) Off J Eur Union L 40:19–25. https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:274:0025:0027:EN:PDF. Accessed 10 Dec 2018
EC Regulation No 183/2005 of the European Parlament and of the Council of 12 January 2005 laying downrequirements of feed hygiene. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32005R0183&from=EN. Accessed 10 Dec 2018
Regulation (EU) 2019/4 of the European Parliament and of the Council of 11 December 2018 on the manufacture, placing on the market and use of medicated feed, amending Regulation (EC) No 183/2005 of the European Parliament and of the Council and repealing Council Directive 90/167/EEC
Commission Decision. 2002/657. Commsision Decision of 12 August 2002 implementing Cuncil Directive 96/23/EC concering the performance of analytical and the interpretation of results. Off Publ Europ Comm 2002, L 221, pp. 8–36
European Medicines Agency EMEA (2006) ICH Topic Q 2 (R1) Validation of analytical procedures: text and methodology
Matuszewski BK, Constanzer ML (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 1:3019–3030
Chou KY, Cheng TY, Chen CM, Hung PL, Tang YY, Chung-Wang YJ, Shih YC (2009) Simultaneous determination of residual thiamphenicol and florfenicol in foods of animal origin by HPLC/electrospray ionization–MS/MS. J AOAC Int 92:1225–1232
Liu H-Y, Lin S-L, Fuh M-R (2016) Determination of chloramphenicol, thiamphenicol and florfenicol in milk and honey using modified QuEChERS extraction coupled with polymeric monolith-based capillary liquid chromatography tandem mass spectrometry. Talanta 150:233–239
Cronly M, Behan P, Foley B, Malone E, Martin S, Doyle M, Regan L (2010) Rapid multi-class multi-residue method for the confirmation of chloramphenicol and eleven nitroimidazoles in milk and honey by liquid chromatography–tandem mass spectrometry (LC–MS). Food Addit Contam A 27:1233–1246
Shinoda N, Kojima F, Sugiura KJ (2011) Simultaneous determination of residues of chloramphenicol and florfenicol in animal feed by liquid chromatography tandem mass spectrometry. J Residuals Sci Tech 8:125–129
Berreto F, Ribeiro C, Bercellos Hoff R, Dalla Costa T (2016) Determination of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in poultry, swine, bovine and fish by liquid chromatography–tandem mass spectrometry. J Chromatogr A 1449:48–53
Dang PK, Degand G, Douny C, Ton V, Maghuin-Rogister G, Scippo ML (2011) Optimisation of a new two-plate screening method for the detection of antibiotic residues in meat. Int J Food Sci Tech 46:2070–2076
Franje CA, Changb SK, Shyua CL, Davisc JL, Leea YW (2010) Differential heat stability of amphenicols characterized by structural degradation, mass spectrometry and antimicrobial activity. J Pharm Biomed 53:869–877
Hayes JM (2005) Determination of florfenicol in fish feed by liquid chromatography. J AOAC Int 88:1777–1783
Hayes JM, Gilewicz R, Freehauf K, Fetter M (2009) Assay of florfenicol in swine feed: interlaboratory study. J AOAC Int 92:340–347
Huet AC, Fodey T, Haughey SA, Weigel S, Elliott C, Delahaut P (2010) Advances in biosensor-based analysis for antimicrobial residues in foods. Trend Anal Chem 29:1281–1294
Luo P, Cao X, Wang Z, Jiang H, Zhang S, Chen X, Wang J, Feng C, Shen J (2009) Development of an enzyme-linked immunosorbent assay for the detection of florfenicol in fish feed. Food Agric Immunol 20:57–65
Luo PJ, Jiang WX, Chen X, Shen JZ, Wu YN (2011) Technical note: development of an enzyme-linked immunosorbent assay for the determination of florfenicol and thiamphenicol in swine feed. J Anim Sci 89:3612–3616
Pouliquen H, Morvan ML (2005) Determination of florfenicol in freshwater, sediments and bryophyte fontinalis antipyretica by HPLC with fluorescence detection. Chromatographia 62:225–231
Shen JZ, Xia X, Jiang H, Li C, Li J, Li X, Ding S (2009) Determination of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in poultry and porcine muscle and liver by gas chromatography–negative chemical ionization mass spectrometry. J Chromatogr B 877:1523–1529
Xie KZ, Jia LF, Yao YL, Xu D, Chen SQ, Xie X (2011) Simultaneous determination of thiamphenicol, florfenicol and florfenicol amine in eggs by reversed-phase high performance liquid chromatography with fluorescence detection. J Chromatogr B 879:2351–2354
Pietroń WJ, Woźniak A, Pasik K, Cybuski W, Krasucka D (2014) Amphenicols stability in mediicated feed—development and validation of liquid chromatography method. Bull Vet Inst Pulawy 58:621–629
SANTE/11813/2017 Guidance document on analytical quality control and method validation procedures for pesticide residues and analysis in food and feed. SANTE/11813/2017
Funding
This work was supported by the National Veterinary Research Institute, Pulawy, Poland.
Author information
Authors and Affiliations
Contributions
Conceptualization and HPLC/MS analysis E.P; writing E.P and K.K.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patyra, E., Kwiatek, K. Quantification and Analysis of Trace Levels of Phenicols in Feed by Liquid Chromatography–Mass Spectrometry. Chromatographia 83, 715–723 (2020). https://doi.org/10.1007/s10337-020-03890-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-020-03890-3