
Abstract
The Squirrel Cuckoo (Piaya cayana) is a widely distributed neotropical species with 14 recognized subspecies. Two parapatric subspecies are distributed in Mexico. P. c. mexicana is endemic to the seasonally dry tropical forests of western Mexico, and P. c. thermophila is found in humid forests from eastern Mexico to western Colombia. The 2 taxa have a small area of overlap on the Isthmus of Tehuantepec, but there is pronounced phenotypic differentiation, and very few reported intermediate individuals between these forms, leading to debate about the taxonomic status of the Mesoamerican taxa. Based on two mitochondrial genes, we used phylogenetic, phylogeographic, morphological, and ecological modelling analyses to describe the evolutionary relationships and paleo-distributional patterns of P. cayana in Mexico. Divergence time estimates suggest that the split between P. c. mexicana and P. c. thermophila occurred during the Pleistocene, about 1.24 Mya. Morphometrics of Mexican subspecies indicate that tail length and the white tail tips are significantly longer in P. c. mexicana, while P. c. thermophila has a longer and wider bill. Ecological niche analyses indicated that niche similarity between both lineages was lower than expected by chance, while low values on cross-prediction tests suggested that the two lineages have inhabited different environmental spaces since at least the Late Pleistocene. The ecological niche difference may also be associated with a steep humidity gradient, suggesting a significant contemporary environmental barrier. Overall, our results indicate that P. c. mexicana and P. c. thermophila have divergent evolutionary histories; therefore, the current taxonomic status of the Piaya populations in Mexico reflects neither their evolutionary relationships nor their apparent divergence. Our results suggest a major split in the polytypic P. cayana coinciding with the Andes, suggesting that the western endemic P. c. mexicana and P. c. thermophila are best treated as separate species-level taxa.
Zusammenfassung
Die Rolle ökologischer und geografischer Faktoren bei der Abstammung und Diversifikation des Eichhornkuckucks Piaya cayana in Mexiko: von der mitochondrialen DNA her betrachtet
Der Eichhornkuckuck (Piaya cayana) ist eine weit verbreitete neotropische Art mit 14 anerkannten Unterarten. In Mexiko gibt es zwei parapatrische Unterarten. P. c. mexicana ist in den je nach Jahreszeit trockenen tropischen Wäldern Westmexikos beheimatet, während P. c. thermophila in den feuchten Wäldern von Ostmexiko bis Westkolumbien vorkommt. Die beiden Taxa überschneiden sich in einem kleinen Gebiet an der Landenge von Tehuantepec, aber es gibt einen ausgeprägten phänotypischen Unterschied und nur sehr wenige bekannte Mischtypen zwischen den beiden und damit Anlass zu Diskussionen über den taxonomischen Status dieser mittelamerikanischen Taxa. Auf der Grundlage zweier mitochondrialer Gene haben wir phylogenetische, phylogeografische, morphologische und ökologische Modell-Analysen durchgeführt, um die abstammungsbiologischen Verbindungen und Verbreitungsmuster von P. cayana im Paläozän in Mexiko zu bestimmen. Schätzungen der Entstehungszeit der Unterschiede deuten darauf hin, dass die Trennung zwischen P. c. mexicana und P. c. thermophila während des Pleistozäns stattfand, vor etwa 1,24 Millionen Jahren. Die morphometrischen Daten der mexikanischen Unterarten zeigen, dass die Schwanzlänge und die weißen Schwanzspitzen bei P. c. mexicana deutlich länger sind, während P. c. thermophila einen längeren und breiteren Schnabel hat. Analysen der ökologischen Nischen zeigten, dass die Übereinstimmungen zwischen den beiden Linien geringer waren als durch Zufall zu erwarten wäre, während niedrige Werte bei Kreuz-Vorhersagetests darauf hindeuteten, dass die beiden Linien mindestens seit dem späten Pleistozän in unterschiedlichen Lebensräumen gelebt haben müssen. Der ökologische Nischenunterschied kann auch mit dem starken Feuchtigkeitsgradienten zusammenhängen, was auf eine bedeutende gegenwärtige Umweltbarriere hinweist. Insgesamt deuten unsere Ergebnisse darauf hin, dass P. c. mexicana und P. c. thermophila eine unterschiedliche Entwicklungsgeschichte haben; daher gibt der derzeitige taxonomische Status der Piaya-Populationen in Mexiko weder ihre evolutionäre Verwandtschaft, noch ihre offensichtlichen Unterschiede wieder. Unsere Ergebnisse deuten darauf hin, dass sich der polytypische P. cayana in den Anden aufgespalten hat, was bedeuten könnte, dass die im Westen endemischen P. c. mexicana und P. c. thermophila am besten als separate Taxa auf Artniveau behandelt werden sollten.
Similar content being viewed by others

Introduction
Variation in phenotypic characters, such as morphology, plumage, behavior, and vocalizations, has been traditionally used as evidence of differentiation, especially at lower taxonomic levels. Phenotypic variation has frequently been the basis for initial taxonomic and evolutionary hypotheses, which are later tested (and either corroborated or contradicted) using molecular analyses (Paterlini 2007; Gill 2014). When populations are divided by conspicuous geographic barriers, genetic differentiation generally coincides with geographic isolation and molecular analyses have frequently supported prior phenotype-based taxonomic proposals (García-Moreno et al. 2004; Navarro-Sigüenza et al. 2008; Barrera-Guzmán et al. 2012; Ortiz-Ramírez et al. 2016). In other cases, genetic variation does not correspond with differentiation patterns based on traditional phenotypic characters or even distributional ranges (Ottvall et al. 2002; Cortés-Rodríguez et al. 2008; Rocha-Méndez et al. 2018). These mixed results have underlined the need to reassess taxonomy in several biotic groups (Fritz et al. 2007; Puebla-Olivares et al. 2008; McCormack et al. 2008).
Geographic barriers are not the only cause of population divergence; divergence can also be promoted or maintained by ecological adaptation to different environments, leading to ecological niche specialization (Moritz et al. 2000; Wiens 2004; Wiens and Graham 2005; Warren et al. 2008). Furthermore, although it is well known that divergence occurs mainly in allopatry (e.g., Zink et al. 2000; Smith et al. 2014; Cracraft et al. 2020), the processes maintaining diversity are less clear (see Peterson et al. 1999), especially in continuous habitats. One example is the continental lowlands, which were temporarily fragmented due to Neogene and Quaternary climatic changes and subsequently rejoined (Weir 2006; Voelker et al. 2010; Smith et al. 2014; Sánchez-González et al. 2015). Recent studies suggest that diversity in Mesoamerican seasonally dry tropical forests has been maintained through persistent but temporarily isolated stable climatic areas (Castillo-Chora et al. 2021). Therefore, allopatry and climatic changes may act in concert at different stages of differentiation; isolation may be the initial catalyst of divergence, which is then further promoted and maintained by habitat dynamics due to climatic changes (Peterson et al. 1999; Hosner et al. 2014; Cabanne et al. 2016; Moreno-Contreras et al. 2020).
The Mesoamerican biogeographic dominion (sensu Morrone 2017) which is included in the Caribbean subregion (Morrone 2001), has been consistently recognized as a biodiversity hotspot in which a high number of species are found in a relatively small area (Myers et al. 2000; Sarkar et al. 2009). The Mesoamerican biogeographic dominion extends from the tropical lowlands of Mexico to northern Nicaragua and is characterized by a highly complex topography, which has promoted the evolution of differentiated populations separated by either lowlands or mountains (Peterson and Navarro 2000; Ornelas et al. 2013; Navarro-Sigüenza et al. 2014; Rocha-Méndez et al. 2019). In most cases, phenotypic differentiation of these populations has been deemed too weak to recognize them as full species (e.g., Howell and Webb 1995; American Ornithologists’ Union 1998), such that variation has mostly been recognized at the subspecies category. The frequency of this practice in ornithology has led to a rampant underestimation of Mesoamerican avian diversity (Navarro-Sigüenza and Peterson, 2004), especially in the isolated dry forests of Mexico’s western Pacific Slope (Peterson and Navarro 2000; Navarro-Sigüenza and Peterson, 2004; Ríos-Muñoz and Navarro-Sigüenza 2012, Navarro-Sigüenza et al. 2014; Prieto-Torres et al. 2019a, b. Recent research has demonstrated that many of these western Mexican bird taxa fulfill criteria for independent species status (e.g., Arbeláez-Cortes and Navarro-Sigüenza 2013; Montaño-Rendón et al. 2015; Vázquez-López et al. 2020), and it is now evident that the region’s ecological and historical dynamics have promoted unexpectedly high levels of differentiation in birds (Castillo-Chora et al. 2021; Sánchez-González et al. 2021) and other vertebrates (e.g., Reyes-Velasco et al. 2013; Arcangeli et al. 2018).
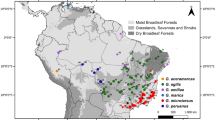
The Squirrel Cuckoo (Piaya cayana) is an arboreal species that is widely distributed in the Neotropics, both geographically (from both coasts in Mexico to northern Argentina) and ecologically (from lowland rainforest to dry forests, gallery forest, mangroves, and areas with scattered trees; Howell and Webb 1995; Payne 1997, 2005). It is also a highly polytypic species, with 14 subspecies recognized based on size, plumage color, and the color of the bare skin around the eye (Sharpe 1900; Ridgway 1916; Peters 1940). Two well-differentiated subspecies are distributed in the Mexican lowlands (Fig. 1). One of them is P. c. mexicana, endemic to the western Pacific slope of Mexico (southern Sonora to southeastern Oaxaca), characterized by lighter cinnamon upperparts and a long tail with a rufous underside. The other is P. c. thermophila which is distributed in forests from northeastern Mexico to western Colombia, characterized by darker cinnamon upperparts and a shorter tail with black underside (Peters 1940; Howell and Webb 1995; Payne 2005). The geographic distribution of these two subspecies overlaps in a small area in the southeastern part of the Isthmus of Tehuantepec (Binford 1989) where there is an ecotone between dry and more mesic environments, suggesting a role of ecological conditions in their divergence. Intergradation between P. c. mexicana and P. c. thermophila is practically unknown. Both forms were initially described as full species (Sharpe 1900), only to be recognized as subspecies within a larger P. cayana complex, because the difference in the color of the upperparts was deemed minor (Stone 1908). Ridgway (1916) later argued that these two forms should be granted full species status based on the absence of intermediate birds in the contact zone. It was not until 70 years later that Binford (1989) reported, without further details, at least four “definite intermediates” individuals from Tehuantepec to Tapanatepec in southeastern Oaxaca. However, Binford himself, wrote “the abruptness and apparent rarity of intergradation suggest that these two forms might be separate species”. Some recent checklists have considered these taxa as subspecies (American Ornithologists’ Union 1998), while others have considered them full species (del Hoyo et al. 2017; Gill Donsker 2017).

Geographic distribution and phylogenetics of the Squirrel cuckoo Piaya cayana (A) mDNA Bayesian phylogenetic tree based on a ND2 (1041 bp) and a fragment of Cytb (622 bp) concatenated dataset. Numbers above branches indicate divergence time (in red), and bootstrap/posterior probability values (in black). The dark gray dotted circle represents the split between South American and Mesoamerican clades, while the light gray circle represents the split between the mexicana and thermophila lineages; B geographic distribution (P. c. thermophila in green, P. mexicana in orange) and mitochondrial DNA population samples (black dots) in Mexico and Central America. Dotted lines depict both the accessible (or M) and projection areas for the models and the niche divergence analyses for the two evolutionary linages. Brown shading indicates areas at least 1000 m above sea level (m.a.s.l); and C haplotype network. Colors correspond to the vertical bar in the phylogenetic tree
Here, we assess the significance of phenotypic differentiation between P. c. mexicana and P. c. thermophila, and the role that ecological factors may have played in the divergence of the two taxa in Mexico. We also assess the phylogenetic and phylogeographic relationships within both groups, to: (1) estimate the divergence time between the two taxa, (2) estimate the genetic diversity and gene flow between them, and (3) examine whether genetic variation may be associated with barriers to gene flow and ecological niche differentiation across the historical distribution throughout the region, with an emphasis in the Isthmus of Tehuantepec.
Materials and methods
Lab procedures and molecular phylogenetic analyses
We used tissue samples and museum skins of P. cayana collected from different localities throughout the distributional range of P. c. mexicana and P. c. thermophila in Mexico (Fig. 1), housed at the Museo de Zoología, Facultad de Ciencias and the Instituto de Biología, of the Universidad Nacional Autónoma de México (UNAM). We supplemented our study with tissue from both Mexico and South America loaned by the Biodiversity Institute, University of Kansas, and the Academy of Natural Sciences of Drexel, both located in the United States. For outgroups, we also used sequences deposited in Genbank (http://www.ncbi.nlm.nih.gov/genbank/).
We extracted DNA from tissue samples using DNeasy kits (Qiagen, Valencia, California) following the manufacturer’s protocols. We amplified two mitochondrial DNA (mDNA) genes, the NADH hydrogenase unit 2 [ND2, 1041 base pairs (bp)] and a fragment of 622 bp from the cytochrome b (Cytb), via PCR (Polymerase Chain Reaction) using standard thermocycle protocols in 12 µl reactions. We used the following primers L5215 (Hackett 1996), H5766 (Sorenson et al. 1999), L5758 (Johnson and Sorenson 1998), 487L (Sánchez-González and Moyle 2011), and H6313 (Johnson and Sorenson 1998) for ND2; and L14996 y H15646 (Sorenson et al. 1999) for Cytb. We visualized PCR products on agarose gels (1.3%) stained with ethidium bromide (0.5 µl/ml). Purification and sequencing were conducted at the High-Throughput Genomics Unit at the University of Washington, Seattle, EUA (http://www.htseq.org/index.html). Sequence chromatograms were edited in Geneious 7.0 (http://www.geneious.com/) and then aligned in MUSCLE (Edgar 2004) using Cytb and ND2 sequences deposited in GenBank. We restricted our analyses to mitochondrial sequences due to limited resources/funds. We are aware that including nuclear genome data can improve the robustness of hypotheses of systematics and gene flow compared to using mitochondrial genes alone (Nichols 2002; Funk and Omland 2003; Ballard and Whitlock 2004; Degnan and Rosenberg 2009); however, mtDNA has been suggested to be reliable for determination of geographical and taxonomic limits in closely related taxa concerning to lineage divergence, population structure, and phylogeograhical patterns (Zink and Barrowclough 2008, and references therein).
For most analyses, we included only individuals for which we sequenced both genes (Supplemental File 1). To better resolve the phylogenetic placement and degree of divergence of the Central American taxa within the polytypic P. cayana, separate analyses containing only Cytb sequences were carried out (Supplemental File 1), given that South American populations are much better represented for this gene on Genbank. We estimated phylogenetic relationships based on a concatenated matrix (ND2 + Cytb gene sequences) and for the Cytb sequences using Maximum Likelihood (ML) as implemented in RAXML 7.0.3 (Stamatakis 2006) and Bayesian Inference (BI) using Mr. Bayes 3.2.2 (Ronquist and Huelsenbeck 2003). Substitution models and partition schemes were estimated separately for ML and BI to increase the accuracy of the phylogenetic inference (Table S1); these were estimated and selected in PartitionFinder 1.1.1 (Lanfear et al. 2012) via the Bayesian Criterion Information (BIC). For Cytb analyses, we used the same models determined for this gene in the concatenated analysis (Table S1). Node support for the ML analyses was assessed using 1 × 104 non-parametric bootstrap iterations. We ran 10 × 106 generations for the BI analysis using four MCMC chains and sampled parameters and trees every 1000 generations. After checking for convergence and stationarity in Tracer 1.6 (Rambaut et al. 2014), the first 3000 trees were discarded as burn-in. The rest of the trees were used to produce a single 50% majority rule consensus tree. Based on a previous higher level phylogenetic hypothesis for the Cuculiformes (Sorenson and Payne 2005), we used Genbank (http://www.ncbi.nlm.nih.gov/genbank/) sequences of the following species as outgroups: Piaya melanogaster, Coccyzus minor, and Coccycua minuta.
Because of our limited sampling of the South American group for the two mitochondrial genes in our analyses, we further tested the monophyly of the three P. cayana groups (see below and Results) using the widespread sampling of South American Cytb sequences in Smith et al. (2014) available in Genbank. We supplemented this mostly South American dataset with our more complete sampling of Mesoamerican individuals and reanalyzed the whole dataset using the same parameters as in Smith et al. (2014).
Divergence time estimations
We estimated divergence time for the Cuculiformes in BEAST 1.8 (Drummond et al. 2012). We used the following substitution rates for our mtDNA markers: 0.029 (0.024–0.033) substitutions/site/lineage/Million years (s/s/l/My) for ND2, and 0.016 (0.014–0.019) s/s/l/My for CytB (Lerner et al. 2011; Lim and Sheldon 2011). Substitution models were specifically estimated for this analysis and selected with a Bayesian Information Criterion (BIC), as implemented in PartitionFinder (Lanfear et al. 2012). Clock model selection for our dataset was performed using Bayes factors log-likelihood values applying a stepping-stone method (Xie et al. 2011) in MrBayes 3.2.2 (Ronquist and Huelsenbeck 2003), which allows the testing of the data for fitting either to a strict molecular clock or a relaxed molecular clock. We ran analyses for 50 × 106 generations, sampling every 1000 generations, and inspected chain convergence in Tracer 1.6 (Rambaut et al. 2014). Stepping-stone analyses favored an uncorrelated relaxed clock (log-likelihood: − 14,511.77) over a strict clock (log-likelihood: − 14,516.36), following Kass and Raftery (1995). We thus implemented a relaxed molecular clock and a birth–death model for the speciation process. As calibration points, we used the most recent common ancestor (MRCA) for Cuculiformes, which has an estimated age of 44 million years (mya), following Prum et al. (2015) and Claramunt and Cracraft (2015), who estimated divergence between Geococcyx and Neomorphus around 7.8 ± 1 mya (Smith and Klicka 2010), and the divergence between Cuculus poliocephalus and the rests of the species in the genus Cuculus around 4.5 mya ± 1 mya (Fossøy et al. 2016). We ran two independent runs for 50 × 106 generations, sampling every 1000 generations; chain convergence was inspected in Tracer 1.6 (Rambaut et al. 2014). Phylogenetic dated trees from the two independent runs were combined using a burn-in of 25% in LogCombiner 1.8 (Drummond et al. 2012), from which we generated a Maximum Clade Credibility Tree in TreeAnotator 1.8 (Drummond et al. 2012).
Phylogeographic analyses
We divided our samples into three groups (Western, Eastern, and South America) for phylogeographic analyses following the topology shown by the phylogenetic trees. As the South American group had a high genetic diversity (Smith et al. 2014), which likely includes several well-differentiated taxa, we included this group only for informative purposes. For each of these groups, we calculated haplotype number (h), nucleotide diversity (π), and haplotype diversity (Hd) as measures of genetic diversity. We also measured the average number of substitutions per site (Dxy) with a Jukes–Cantor correction and the net number of substitutions per site (Da) as measures of genetic differentiation. All these statistics were estimated in DNAsp 5.1 (Librado and Rozas 2009).
We also explored the genetic structure within P. cayana using a three-way AMOVA on the previously described groups. The significance of the AMOVA results was assessed through 10,000 non-parametric permutations. In addition, we estimated Fst as a measure of population structure (Weir and Cockerham 1984), which take values ranging from panmixia (0) to a complete partition of genetic variation among subpopulations (1) following Hamilton (2009). We also assessed the significance of the Fst parameter with 10,000 permutations. Both AMOVA and Fst analyses were conducted in Arlequin 3.5.1.3 (Excoffier and Lischer 2010).
Since derived haplotypes may coexist with ancestral haplotypes within a single population, it has been suggested that there may be reticulated relationships that are not represented in bifurcated intraspecific phylogenetic trees (Posada and Crandall 2001). Haplotype networks are thus an additional option for representing these reticulated patterns. We constructed haplotype networks for the concatenated and the Cyb analysis based on the statistical parsimony algorithm implemented in PopArt 1.7 (Leigh and Bryant 2015).
Morphometrics
We measured eight morphometric characters from 110 P. cayana study skins from Mexico housed in scientific collections (Museo de Zoología of the Facultad de Ciencias and the Colección Nacional de Aves of the Instituto de Biología, both at the Universidad Nacional Autónoma de México). The selected characters are likely related to habitat use, foraging (Miles and Ricklefs 1984; Jønsson et al. 2015) and communication in Cuculiformes (Payne 1997) and are considered reliable indicators for size and the least prone to deformation during the skinning process (Zink and Remsen 1986). We measured the following characters: bill length (BL), width (BW), and depth (BD), all taken at the distal end of the nostril; wing chord (WC); and the lengths of the tarsus (TL), hallux (HAL), tail (TAIL), and of the white tips at the end of the underside of the tail (TIPS). Most measurements were taken with a Mitutoyo CD-6″ digital caliper to the nearest 0.01 mm, except for WC and TAIL, for which we used a-50 cm ruler (accurate to the nearest 1 mm). We ensured the repeatability of measurements by measuring a series of 10 specimens chosen at random during three different sessions on consecutive days until Pearson correlation tests showed only positive and significant correlations (Benítez-Díaz 1993). We measured only adult male specimens to avoid variation due to age (Yezerinac et al. 1992) and sex (Holmes 1986).
As we measured only Mexican specimens, for all subsequent statistical analyses, specimens included only representatives of the Western lineage (cinnamon undertail feathers, from the Pacific slope of western Mexico) and the Eastern lineage (black undertail feathers, from the Gulf Slope and the Yucatan Peninsula). We considered Pearson’s correlation coefficient values > 0.75 between measured characters to indicate redundancy (Zink and Remsen 1986; Rising and Somers 1989). This threshold was not met for any of the pairs of measured characters, so we included all variables measured for all subsequent analyses. We tested for significant differences in morphometric differences between the two lineages using a Mann–Whitney U test (Fay and Proschan 2010) and visualized these differences using box and whiskers plots. We conducted all these analyses in R (R Core Team, 2014). Some measurements were absent because of damaged specimens (e.g., lacking tail feathers). We therefore used a probabilistic Principal Components Analysis (PPCA), as implemented in pcaMethods (Stacklies et al. 2007) for R (R Core Team 2014) to allow the inclusion of individuals with missing data.
Paleodistributions and niche divergence analyses
To assess the variation of geographical and environmental conditions throughout the distribution of P. cayana in Mexico, we used ecological niche modelling (ENM) to approximate the Grinnellian niches (Soberon and Peterson 2005) occupied by each lineage. Georeferenced records in these analyses corresponded to specimens used in the morphometric and genetic analyses as well as occurrence records from P. cayana museum specimens included in the Global Biodiversity Information Facility database (GBIF.org 2021; Supplemental Materials I). To correct for spatial bias and possible correlated localities due to a high density of collected specimens (see Peterson et al. 2011), we retained only one presence point (i.e., an independent locality) within a buffer of ~ 7 km2 (the mean distance among occurrence records). We also omitted records that lacked bioclimatic data (see below). The final spatially rarefied dataset consisted of 175 records for P. c. mexicana and 545 for P. c. thermophila. We also defined an accessibility area (or “M” according to the BAM diagram; Soberon and Peterson, 2005; Barve et al. 2011) for each evolutionary lineage, based on occurrence records matching the terrestrial ecoregions (Dinerstein et al. 2017) and the biogeographic provinces of the Neotropical region (Morrone 2014). We assumed that these regions define the historically accessible area for each lineage in the geographical space (Svenning and Skov 2004). To characterize the ecological niche of the two lineages, we used nine uncorrelated variables at a spatial resolution of 0.041665° (~ 5 km2) obtained from WorldClim 1.4 (Hijmans et al. 2005): mean diurnal range (Bio 02), annual temperature range (Bio 07), mean temperature of the wettest quarter (Bio 08), annual precipitation (Bio 12), precipitation of wettest month (Bio 13), precipitation of driest month (Bio 14), precipitation seasonality (Bio 15), precipitation of driest quarter (Bio 17), and precipitation of coldest quarter (Bio 19). The selection of these climatic variables was based on the Pearson’s correlation coefficient (r < 0.8) and Variance Inflation Factor (VIF < 10), as implemented in the “corrplot” (Wei and Simko 2017) and “usdm” (Naimi 2015) R libraries.
Using MaxEnt 3.4.1 k (Phillips et al. 2006) and the “kuenm” R package (Cobos et al. 2019), we defined ENMs representing the spatio-temporal potential geographic distribution during the Late Pleistocene climate fluctuations, which allowed us to identify the potential historical range extent, connectivity, and stability areas for each lineage. To do this, we applied a four-step methodological approach (see Peterson et al. 2011): (1) calibration of ENM for each lineage in the present (hereafter Recent) to assess the model complexity and select the best modelling parameters (see Cobos et al. 2019); (2) selection and validation of models in terms of omission (under-prediction) errors, the partial-ROC test, and AIC values (Anderson et al. 2003; Peterson et al. 2008; Cobos et al. 2019); (3) projection of the defined suitable environmental conditions to climatic conditions during the Mid-Holocene (Mid-Hol, 6000 Ya), and the Late Pleistocene climate fluctuations including the Last Glacial Maximum (LGM; 21,000 Ya) and Last Inter Glacial (LIG, ~ 120,000–140,000 Ya); and (4) conversion of the logistic suitability values into a binary (presence–absence) map by setting a decision threshold equivalent to the 10th percentile training presence logistic threshold (see Liu et al. 2013, 2016). Past climate data were extracted from three global climate circular models: CCSM4 (Collins et al. 2004), MIROC-ESM (Hasumi and Emori 2004), and MPI-ESM-P (Baehr et al. 2015). We tested the reliability of our model projections by calculating the mobility-oriented parity (MOP) metric (Owens et al. 2013), which offers a view of the novelty of past climate conditions relative to present-day conditions within the calibration area for each lineage (see Alkishe et al. 2017 for a complete explanation). Detailed descriptions of the methods used to determine ENMs and MOP for each lineage are provided in Supplemental File II. All scripts and input information are available at https://github.com/davidprietorres/piaya_cayana_paleodistribuion.
To understand the role of geographical drivers of lineage diversification in P. cayana in Mesoamerica, we assessed the degree of geographical matching between the potential ranges for the different lineages predicted across each climatic scenario (i.e., alloprediction values). Thus, models are expected to predict similar potential distribution ranges if the climatic niche for lineages is similarly distributed and historically congruent despite geographical barriers (Mota-Vargas and Rojas-Soto 2016). Also, we obtained long-term climatic stability areas or refugia for each lineage by overlapping the binary maps obtained for each climatic scenario (Carnaval et al. 2009); a cell or pixel is considered climatically stable if lineages are predicted to occur there in all of the historical scenarios analyzed (Terribile et al. 2012). This procedure allows the analysis of the significance of climatic stability/instability areas in the evolution and maintenance of lineage diversity (e.g., Carnaval and Mortiz 2008; Carnaval et al. 2009; Cabanne et al. 2016; Castillo-Chora et al. 2021).
We also estimated the climate niche overlap between the lineages using the PCA-env method (Broennimann et al. 2012). We used a principal component analysis (PCA) to transform the environmental space of the selected environmental variables into a two-dimensional space defined by the first and second principal components (Strubbe et al. 2015). We then calculated the niche overlap between linages directly from the ecological niche space (Warren et al. 2008) using the mean of Schoener’s D, which ranges from 0 (no niche overlap) to 1 (complete niche overlap). We compared this value with randomly generated simulated values from “niche identity test” and “background similarity test” metrics (see Warren et al. 2008; Broennimann et al. 2012). The hypothesis of niche conservatism between lineages is accepted only if the observed Schoener’s D value is significantly higher (P < 0.05; i.e., more similar) than values expected for simulated overlap from the 1,000 pseudo-replicated data sets allowing random shifts. All analyses were estimated in “ecospat v. 3.0” (Di Cola et al. 2017) for R (R Core Team, 2014). Finally, we used linear and blob range-break tests to test whether the geographical/ecological boundaries between the Western and Eastern lineages may be associated with steep environmental gradients, as implemented in ENMTools 1.0 (Warren et al. 2021) for R (R Core Team, 2014). We ran 100 pseudoreplicates for each test to generate a null distribution, which we compared to the observed overlap value via a one-tailed test (see Glor and Warren 2011).
Results
Phylogenetic analyses
We obtained 32 ND2 and Cytb sequences from 26 localities (1–4 samples per locality) distributed across the geographic range of P. cayana (Fig. 1, Supplemental File 1). Sequences are deposited in Genbank, under accession numbers OM169114-OM169145 for ND2, and OM169146-OM169177 for Cytb.
Phylogenetic trees obtained for both ML and BI from the concatenated dataset showed highly similar topologies; in both cases, two well-supported main clades were recovered (Fig. 1A): a Mesoamerican clade grouping all samples from Mexico and Central America was sister to a monophyletic group including all samples from South America (South American clade). The Mesoamerican clade was further subdivided into two reciprocally monophyletic and well-supported clades: one grouped all samples from the western Pacific Slope of Mexico (Western lineage), and a second group including all samples from the Gulf Slope, the Yucatan Peninsula, and Central America (Eastern lineage). The geographic structure observed in these two Mesoamerican clades matches the distribution of the two recognized Mexican subspecies P. c. mexicana and P. c. thermophila. However, the phylogenetic tree derived from complementary analysis based on the Cytb dataset revealed that the Chocoan P. c. nigricrissa was placed with marginal support (PP = 0.72) as sister of the Mexican endemic P. c. mexicana (Supplemental Fig. 1). Results of our overall analyses based on both the concatenated and Cytb trees outline the significance of the Andes as a barrier, and therefore, a Trans-Andean clade (including P. c. mexicana, P. c. thermophila and the Chocoan P. c. nigricrissa) and a Cis-Andean clade (including all South American samples in this work) might be recognized. In addition, both datasets also showed two clades with Mesoamerican P. cayana, supporting a clear split between the western endemic P. c. mexicana and the Gulf slope-Central American P. c. thermophila.
Phylogeographic analyses
The three studied clades showed high Hd values (> 0.8), lower values for nucleotide diversity (π < 0.2), and a large number of haplotypes (Table S2). Genetic differentiation among lineages based on the concatenated dataset is deep, with Dxy values for the genetic distance between the Mesoamerican clade and the South American clade higher than 3%. In comparison, the genetic distance between the Western and Eastern lineages is 2.4% (Table 1). According to the Cytb dataset analysis, the Dxy value between the Trans-Andean clade and the Cis-Andean clade was 3.1%, while Dxy values for the Western lineage and the Chocoan nigririssa were of 2.6%; of 3.1% between the Eastern and Western lineages; and of 2.8% between the Eastern lineage and nigririssa. The AMOVA analysis revealed that most of the genetic variation (73.14%) is distributed between the Eastern and Western lineages within the Mesoamerican clade. In comparison, a non-significant portion of the variation (17.27%) is distributed between the Mesoamerican and the South American lineages (Table 2).
The haplotype network showed a clear geographic structure matching the clades in the phylogenetic trees (Fig. 1C). No haplotypes were shared between different lineages, and we detected large numbers of mutational steps separating the different groups; 32 mutational steps separated the South American clade from the Mesoamerican clade; and within the Mesoamerican clade, 35 mutational steps and a hypothetical haplotype separated the Western and the Eastern lineages (Fig. 1C). A star-like pattern is evident in the Eastern lineage. We detected no clear pattern in the case of the Western lineage, for which several hypothetical or unsampled haplotypes were determined. The inclusion of the single Chocoan sequence showed a haplotype network in which this sample was also placed as closer to the Western lineage, within a Trans-Andean clade (Supplemental Fig. 2).

Morphometrics for the two Mesoamerican lineages. Orange color depicts mean and samples for the Western lineage (P. mexicana); green color indicates mean and samples for the Eastern lineage (P. c. thermophila). Mann–Whitney U test boxplots for comparison of the means for the different characters. Abbreviations: Bill width (BW), Bill Length (BL), Bill Depth (BD), Tail Length (TAIL), Length of white tips in the tail (TIPS), Hallux Length (HAL), Tarsus Length (TL), and Wing Chord (WC). Right: principal component analysis plot showing the distribution of individuals in each lineage in the morphological space
Divergence time
Our results suggest that the South American and the Mesoamerican clades diverged during the Late Miocene–Early Pliocene, about 4.7 mya (6.5–3.2 mya, 95% HPD). Within the Mesoamerican clades, the Western and the Eastern clades diverged around 1.24 mya (1.8 – 0.8 mya, 95% HPD), during the Early Pleistocene (Fig. 1).
Morphometrics
We measured a total of 94 study skins (Supplemental File I). Boxplots for univariate analyses showed similar measurements between the lineages for all of the characters, except for TAIL (Fig. 2). The Mann–Whitney U test showed significant differences in TAIL, bill length (BL), and bill width (BW). The Western lineage (P. c. mexicana) had a significantly longer tail and shorter and narrower bill than the Eastern lineage (P. c. thermophila).
Pearson correlation tests showed no significant correlations between pairs of morphological measurements (Table S3), so we included all of the variables in the PCA. The first three components of the PCA explained 83.44% of the total variation: 58.24% in PC1, 19.02% in PC2, and 6.18% in PC3 (Table S4). For PC1, significant loadings were concentrated in TIPS and TAIL, while for the PC2, significant loadings were detected for BL and BW. Plotting individual values of the first two components suggests separation into two phenotypic groups corresponding to the Eastern and Western lineages (Fig. 2).
Distribution modelling and paleodistribution
ENMs yielded a good fit for Recent distribution for both the Western (partial-ROC test = 1.34; omission rate = 3.5%; AICc = 2754.29) and the Eastern (partial-ROC test = 1.16; omission rate = 4.6%; AICc = 9360.96) lineages. These results indicate a high model performance, showing that models are better than random and thus statistically descriptive of the climatic conditions defining the ecological niche of each lineage. According to the Jackknife test and contribution variables values in MaxEnt, the most important variables for model building for the Western lineage were Mean Diurnal Temperature Range (Bio 02, 25.8%), Temperature Annual Range (Bio 07, 22.9%), and Precipitation seasonality (Bio 15, 17.3%). For the Eastern lineage, the most important variables were Mean Diurnal Temperature Range (Bio 02, 32.2%), Annual Precipitation (Bio 12, 18.6%), and Precipitation of the Wettest Month (Bio 13, 16.4%).
Mean MOP values for the three past periods suggest that the presence of non-analogous climate areas is not a factor driving the interpretation of our predictions under past environmental conditions (Supplemental File II). Paleodistribution models (Fig. 3) for the Western lineage showed a decrease (~ 12%) in high suitability areas during the LGM compared to Recent, while the suitable area for the Eastern lineage was larger (about 45%) and less fragmented. Overall, predicted potential ranges for both lineages during the LGM showed a lower overlap (on average 54.35%) than predictions for the LIG (77.89%) and mid-Holocene (79.57%) in comparison to their corresponding Recent climate predictions. Alopredictions (i.e., predicted distributions of the Eastern lineage into the range of the Western lineage and vice versa) showed 24.29% overlap, averaging across the four scenarios (30.78% for the Recent, 21.56% for the mid-Holocene, 15.52% for the LGM, and 29.28% for the LIG), suggesting that environmental differentiation has been maintained since at least the Late Pleistocene. Our results showed that the distribution of suitable areas for the two lineages was highly fragmented and ecologically differentiated during the LGM, with areas of suitable habitat separated by the Isthmus of Tehuantepec, unlike Recent, mid-Holocene, and LIG predictions (Fig. 3). Predicted areas of climatic stability for each lineage (crossed areas within the Recent distribution map, Fig. 3) covered 38.62% of the Recent distribution for the Western lineage and 52.56% for the Eastern lineage, showing an overlap of 10.55% and 5.15% with the predicted potential ranges, respectively.

Ecological niche model projected onto the geographic areas for P. mexicana (orange) and P. c. thermophila (green). Upper maps depict suitability areas for each lineage under Recent climatic conditions; crossed lines depict regions of historical climatic stability. Lower maps depict the predicted regions of historical distribution for climatic niches across four time periods, and the aloprediction areas (darker orange and darker green) between lineages across the Late Pleistocene (LIG and LGM), the Holocene and the Recent. Bars in the upper right correspond to the size of the areas predicted by models for each lineage, and their consensus areas (blue color) across the four paleoclimate scenarios
Ecological niche similarity analyses
The PCA-env indicated that 80.11% of the environmental variation is explained by the two first principal components (PC1 = 66.05% and PC2 = 14.06%). The occurrence density surfaces (black spots) in environmental space, as determined by PCAenv, showed a clear difference in position between the lineages (Fig. 4A). Schoeners’ D observed values were 0.09 and statistically non-significant in both directions (P = 0.17, from P. c. mexicana to P. c. thermophila; and P = 0.52, from P. c. thermophila to P. c. mexicana), indicating that the observed niche was no more similar than expected by chance (Fig. 4B, C). This allowed us to reject the hypothesis of niche equivalency and similarity (i.e., niche conservatism) between lineages, suggesting that lineages occupy different environmental niches. Finally, the linear and blob range-break tests detected significant environmental differences (P < 0.05) separating the Western and Eastern lineages.

Equivalence and similarity test in the environmental space for Squirrel cuckoo Piaya cayana lineages in Mesoamerica. A Principal Component Analysis (PCA) of ecological niche for the lineages and the corresponding values of variables contribution. Grey shading in the upper right plots depicts the density of the occurrences of each lineage by cell in the ecological space; the solid and dashed lines indicate 100 and 50% of the available (background) environment, respectively. B Plots of the similarity test comparing the ecological niche of the two lineages in both directions (P. mexicana vs. P. c. thermophila and vice versa). We only present values for the D metrics for both tests. In all plots, the arrow with a red diamond represents the observed similarity value between niches; the gray columns represent 1000 randomly simulated expected values. The P value is shown in each plot (n.s non-significant)
Discussion
Our phylogenetic analyses based on the concatenated dataset showed the existence of two main clades within P. cayana. One clade grouped all samples distributed in Mesoamerica, while the second grouped all samples from South America. The Mesoamerican clade was further subdivided into two reciprocally monophyletic groups divided by the Isthmus of Tehuantepec in southern Mexico: P. c. mexicana to the west and south and P. c. thermophila to the north and east. The pattern of differentiation into two clades is congruent with the geographic limits previously established for the two currently accepted subspecies (Ridgway 1916; Peters 1940), and presumably corresponds with contemporary ecological barriers set by a humidity gradient in the Isthmus of Tehuantepec region (Binford 1989). In addition, our complementary Cytb analyses also showed these Mesoamerican and South American clades, but placed the Chocoan sample as sister to the endemic P. c. mexicana, although with marginal support. The placement of Chocoan nigricrissa within the Mesoamerican clade corroborates the recognition of the Andes as the main for the diversification in P. cayana. The inclusion of nigricrissa in a broader Trans-Andean clade and sister to mexicana further supports the evolutionary divergence of the Western lineage, and may even support the species-level recognition of P. thermophila. Further analyses based on both more specimens and complete genomic datasets are needed to clarify the phylogenetic position of the Chocoan population of the polytypic P. cayana.
Some phylogeographic studies of montane birds (see Barber and Klicka 2010) and other biological groups (Ornelas et al. 2013) have suggested multiple pulses of diversification across the Isthmus of Tehuantepec, in which some of the co-distributed taxa have experienced either simultaneous or different divergence events. These studies have dated some pulses in the Early Pleistocene (1.8–0.78 mya), coinciding with our estimates of divergence between the Western and the Eastern lineages in Mesoamerica. The probable simultaneous divergence for biotas across the Isthmus of Tehuantepec (based on the overlap in the confidence intervals) suggests a vicariant event that limited gene flow for both montane and lowland populations on both sides of the Isthmus (see Miller et al. 2005). This vicariant pattern is relatively well understood in montane populations, for which an east-to-west differentiation axis has been observed (Sullivan et al. 2000; Barber and Klicka 2010; Ornelas et al. 2013). However, it is still poorly understood in lowland birds, in which both east–west and north–south axes have been detected (e.g., Phillips 1986; Binford 1989; Ríos-Muñoz and Navarro-Sigüenza 2012; Prieto-Torres et al. 2019a, b; Mendonça et al. 2021). Differentiation along these axes in this region has been related to a possible seaway across the southern Isthmus of Tehuantepec that may have affected both lowland and highland vertebrates with different dispersal capabilities. However, this theory has been challenged (Mulcahy et al. 2006; Barber and Klicka 2010; but see Durham et al. 1955; Whitmore and Stewart, 1965; Miller et al. 2005); opening the way for alternative explanations. Thus, ecological differentiation has become a natural subject of research (see Hernández-Canchola and León-Paniagua 2017; Hernández-Romero et al. 2018; Moreno-Contreras et al. 2020).
Several aspects of our results support genetic differentiation between the lineages (i.e., genetic distances, haplotype network, and high levels of Fst), which indicates the presence of a long-lasting vicariant barrier that maintained the differentiation of these taxa. Genetic divergence in isolation in Piaya may have been reinforced by low population sizes, as well as by presumably low dispersal capabilities. This is suggested by the relatively short and rounded wings and the extensive geographic variation throughout the apparent habitat continuity across the Neotropics (Payne 2005; Smith et al. 2014).
Morphometrics
Morphological traits may reveal relationships between organisms and their environments (Hedenström 2008; Jønsson et al. 2015). Considering that both lineages inhabit environmentally different regions (Fig. 4), bill traits may differ between the lineages in a response to differences in the prey consumed, which include insects and small vertebrates (Raw 1997; Piratelli and Pereira 2002; Komar and Thurber 2003; Azevedo Silva et al., 2006; Repenning et al. 2009). These preys are from distinctive biotic assemblages in the lowlands of the two slopes. We also detected significant differences in the tail and tail color patterns between P. c. thermophila and P. c. mexicana. These two traits are important in communication and interspecific recognition among cuckoos (Payne 2005). The tail display is presumed to be a critical element of courtship, especially for members of the subfamily Coccyzinae, to which Piaya belongs. The contrast between the tail underside and the white tail tips presumably plays a significant role (Payne 1997). In addition, the color of the underside of the tail seems to be associated with sexual maturity, being pale and less defined in juveniles (Howell and Webb 1995; Payne 2005). Given the significance of the tail in communication and sexual behavior, differences in the tail size and the color of the tail underside may represent prezygotic barriers between the P. c. thermophila and P. c. cayana lineages.
Niche differentiation and geographical barriers
The background similarity test results suggest that the environmental niches occupied by the Eastern and Western lineages are not more similar than expected by chance. This result matches findings in other co-distributed taxa (e.g., Vázquez-Miranda et al. 2009; Arbeláez-Cortes and Navarro-Sigüenza, 2013; Hernández-Canchola and León-Paniagua 2017; Vázquez-López et al. 2020). Since divergence between lineages occurred during the Middle Pleistocene, the final uprising of the Trans Mexican Volcanic Belt may have led to drier conditions across the Pacific Slope (Graham and Dilcher 1995; Becerra 2005; Pennington et al. 2000; Molnar and Cane 2007). These drier environments may have enhanced the adaptation of Piaya populations, leading to geographic restriction and the evolution of many endemic lineages extended across the Pacific Slope tropical dry forests (Becerra 2005; Ramírez-Barrera et al. 2018; Prieto-Torres et al. 2019a, b; Castillo-Chora et al. 2021).
Models under past environmental conditions suggest that during the Quaternary, the Eastern lineage experienced a stronger reduction in high suitability areas than the Western lineage, which has broader habitat suitability areas. Although environmental stability areas for these two taxa were presumably extensive, there seems to have lower connectivity for the Eastern lineage. Furthermore, there appears to have been little-to-no overlap between the stability areas for each lineage, including in the small area where the two lineages currently overlap in the Isthmus of Tehuantepec (see Fig. 3), which may explain the scarcity of intermediate birds. Because ecological niche tests rejected the niche conservatism hypothesis, it is plausible that the two lineages have inhabited different environmental spaces since at least the Late Pleistocene. Our analyses suggest that, although the Isthmus of Tehuantepec has been most widely recognized as a biogeographic limit for different montane taxa in several vertebrate groups (Sullivan et al. 2000; Barber y Klicka 2010; Ornelas et al. 2013; Rodríguez‐Gómez et al. 2013; Ortiz-Ramírez et al. 2016), lowland taxa have also responded to this biogeographic boundary, likely through the evolution and maintenance of ecological niche differences, as we found in Mesoamerican Piaya.
Our analyses of niche divergence, morphometrics, and geographic range suggest that P. c. thermophila and P. c. mexicana are reproductively isolated. However, this will need further corroboration with nuclear and genomic analyses to fully detect reduction or lack of gene flow. Our results using mitochondrial genes provide evidence of a little-to-no gene flow at a single locus; this does not rule out substantial gene flow in the nuclear genome, as has been found by several studies showing discrepancies between the mitogenome and the nuclear genome (Nichols 2002; Funk and Omland 2003; Ballard and Whitlock 2004; Degnan and Rosenberg 2009). Thus, studies considering the entire genome or at least a substantial part of it will be needed to fully corroborate a lack of gene flow.
Systematics
Although the taxonomic debate about the P. cayana complex has been active, to date, it has mostly relied only on phenotypic characters and geographic distribution. However, the study of Smith et al. (2014) based on the Cytb mitochondrial gene suggested highly structured genetic diversity, with four genetically distinct lineages in South America and at least one in Mesoamerica. In that study, Mesoamerica was represented only by samples of thermophila; thus, relationships among taxa within Mesoamerica were not explored.
Our findings on phylogenetics, gene flow, morphometrics, and ecological niche differentiation, together with previously described differences in coloration patterns (Stone 1908; Ridgway 1916), all support the evolutionary divergence of the thermophila and mexicana lineages. The scarcity of proven intermediate individuals or hybrids despite the existence of a (relatively small) contact zone in the southeastern Isthmus of Tehuantepec may be explained by a lack of gene flow and presumably environmental niche differences. In addition, the Eastern and Western lineages show morphological differences in tail traits that are likely involved in communication and reproductive behavior (Payne 2005), and therefore may contribute to reproductive isolation. Evidence from all of these analyses suggests that the continued treatment of the Mexican lineages of P. cayana as a single species (e.g., Peters 1940; American Ornithologists’ Union, 1998) does not accurately reflect the evolutionary divergence of these groups in Mesoamerica. Our mtDNA phylogenetic trees suggest that both P. c. mexicana and P. c. thermophila should be recognized as a separate species from P. cayana (del Hoyo et al. 2017; Gill Donsker, 2017). Furthermore, the placement of the Chocoan nigricrissa within the Trans-Andean clade underscores the need of a more complete sampling of both P. c. thermophila and P. c. nigricrissa in southern Central America and the Chocó region before this can be determined. Our Cytb tree suggest that the monophyly of P. c. thermophila still needs to be proven, due to the close geographic proximity of Piaya c. nigricrissa in the Ecuadorian Pacific Slope of the Andes (Stone 1908).
As evidenced by our set of analyses, a provisional but potentially more accurate taxonomic scheme for Piaya would be the recognition of both mexicana and thermophila (and perhaps nigricrissa) as independent evolutionary lineages that may be recognized under different species concepts (Cracraft 1983; De Queiroz 2007; Gill 2014). Estimates of genetic and divergence times in these lineages are similar or even greater than those found in widely recognized full species of cuckoos or other bird groups (e.g., Johnson and Cicero 2004; Fossøy et al. 2016).
Conclusions
Our phylogenetic analyses suggest that Mexican populations of P. cayana represent at least two separate lineages, which are independent from Cis-Andean South American lineages (see Smith et al. 2014). Each Mesoamerican lineage shows diagnostic characters, and several lines suggest highly reduced gene flow. Estimated divergence dates indicate that the split between thermophila and P. mexicana occurred during the Pleistocene (0–78–1.79 mya). The decrease in habitat extent and connectivity promoted by Pleistocene climatic variation may have enhanced the geographic isolation pulses and the evolution of morphometric differences, which are probably involved in reproduction behavior, which may have also prevented gene flow.
Availability of data and materials
All scripts and input information for ecological analyses are available in GitHub (https://github.com/davidprietorres/piaya_cayana_paleodistribuion).
Code availability
Not applicable.
References
Alkishe AA, Peterson AT, Samy AM (2017) Climate change influences on the potential geographic distribution of the disease vector tick Ixodes ricinus. PLoS ONE 12:e0189092. https://doi.org/10.1371/journal.pone.0189092
American Ornithologists’ Union (1998) Check-list of North American birds, 7th edn. American Ornithologists’ Union, Washington, D.C
Anderson RP, Lew D, Peterson AT (2003) Evaluating predictive models of species’ distributions: criteria for selecting optimal models. Ecol Modell 162:211–232. https://doi.org/10.1016/S0304-3800(02)00349-6
Arbeláez-Cortes E, Navarro-Sigüenza AG (2013) Molecular evidence of the taxonomic status of western Mexican populations of Phaethornis longirostris (Aves: Trochilidae). Zootaxa 3716:81–97. https://doi.org/10.11646/zootaxa.3716.1.7
Arcangeli J, Light JE, Cervantes FA (2018) Molecular and morphological evidence of the diversification in the gray mouse opossum, Tlacuatzin canescens (Didelphimorphia), with description of a new species. J Mammal 99:38–158. https://doi.org/10.1093/jmammal/gyx173
de Azevedo Silva PL, Neto AF, Ferreira AA (2006) Análise do conteúdo estomacal das aves da coleção científica da Universidade Federal de Goiás/Universidade Estadual de Goiás.IV Seminário de Iniciação Científica, Brasil.
Baehr J, Fröhlich K, Botzet M, Domeisen DI, Kornblueh L, Notz D, Pointek R, Pohlmann H, Tiestche S, Müller WA (2015) The prediction of surface temperature in the new seasonal prediction system based on the MPI-ESM coupled climate model. Clim Dyn 44:2723–2735. https://doi.org/10.1007/s00382-014-2399-7
Ballard JW, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13:729–744
Barber BR, Klicka J (2010) Two pulses of diversification across the Isthmus of Tehuantepec in a montane Mexican bird fauna. Proc R Soc Lond B Biol Sci 277:2675–2681. https://doi.org/10.1098/rspb.2010.0343
Barrera-Guzmán AO, Milá B, Sánchez-González LA, Navarro-Sigüenza AG (2012) Speciation in an avian complex endemic to the mountains of Middle America (Ergaticus, Aves: Parulidae). Mol Phylogenet Evol 62:907–920. https://doi.org/10.1016/j.ympev.2011.11.020
Barve N, Barve V, Jiménez-Valverde A, Lira-Noriega A, Maher SP, Peterson AT, Villalobos F (2011) The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol Modell 222:1810–1819. https://doi.org/10.1016/j.ecolmodel.2011.02.011
Becerra JX (2005) Timing the origin and expansion of the Mexican tropical dry forest. PNAS 102:10919–10923. https://doi.org/10.1073/pnas.0409127102
Benítez-Díaz H (1993) Geographic variation in coloration and morphology of the Acorn Woodpecker. Condor 95:63–71. https://doi.org/10.2307/1369387
Binford LC (1989). A distributional survey of the birds of the Mexican state of Oaxaca. Ornithol Monogr. 43
Broennimann O, Fitzpatrick MC, Pearman PB, Petitpierre B, Pellissier L, Yoccoz NG, Thuiller W, Fortin MJ, Randin C, Zimmermann NE, Graham CH, Guisan A (2012) Measuring ecological niche overlap from occurrence and spatial environmental data. Glob Ecol Biogeogr 21:481–497. https://doi.org/10.1111/j.1466-8238.2011.00698.x
Cabanne GS, Calderón L, Trujillo Arias N, Flores P, Pessoa R, d’Horta FM, Miyaki CY (2016) Effects of Pleistocene climate changes on species ranges and evolutionary processes in the Neotropical Atlantic Forest. Biol J Linn Soc Lond 119:856–872. https://doi.org/10.1111/bij.12844
Carnaval AC, Moritz C (2008) Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35:1187–1201. https://doi.org/10.1111/j.1365-2699.2007.01870.x
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323:785–789. https://doi.org/10.1126/science.1166955
Castillo-Chora VDJ, Sánchez-González LA, Mastretta-Yanes A, Prieto-Torres DA, Navarro-Sigüenza AG (2021) Insights into the importance of areas of climatic stability in the evolution and maintenance of avian diversity in the Mesoamerican dry forests. Biol J Linn Soc Lond 132:741–758. https://doi.org/10.1093/biolinnean/blaa202
Claramunt S, Cracraft J (2015) A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci Adv 1:e1501005. https://doi.org/10.1126/sciadv.1501005
Cobos ME, Peterson AT, Barve N, Osorio-Olvera L (2019) kuenm: an R package for detailed development of ecological niche models using Maxent. PeerJ 7:e6281. https://doi.org/10.7717/peerj.6281
Collins WD, Blackmon M, Bitz C, Bonan G, Bretherton CS (2004) The community climate system model: CCSM3. J Clim 19:2122–2143. https://doi.org/10.1175/JCLI3761.1
Cortés-Rodríguez N, Hernández-Baños BE, Navarro-Sigüenza AG, Peterson AT, García-Moreno J (2008) Phylogeography and population genetics of the Amethyst-throated Hummingbird (Lampornis amethystinus). Mol Phylogenet Evol 48:1–11. https://doi.org/10.1016/j.ympev.2008.02.005
Cracraft J (1983) Species concepts and speciation analysis. In: Johnston F (ed) Current ornithology, vol 1. Springer, New York, pp 159–187
Cracraft J, Ribas CC, d’Horta FM, Bates J, Almeida RP, Aleixo A, Boubli JP, Campbell KE, Cruz FW, Ferreira M, Fritz SC, Grohmann CH, Latrubesse EM, Lohmann LG, Musher LJ, Nogueira A, Sawakuchi AO, Baker PA (2020) The origin and evolution of Amazonian species diversity. In: Rull V, Carnaval A (eds) Neotropical Diversification: Patterns and Processes. Springer, Cham, pp 225–244
De Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56: 879-886. https://doi.org/10.1080/10635150701701083
Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24:332–340
del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E (eds) (2017) Handbook of the birds of the world alive. Lynx Edicions, Barcelona
Di Cola V, Broennimann O, Petitpierre B, Breiner FT, D’Amen M, Randin C, Engler R, Pottier J, Pio D, Dubuis A, Pellissier L, Mateo RG, Hordijk W, Salamin N, Guisan A (2017) ecospat: an R package to support spatial analyses and modeling of species niches and distributions. Ecography 40:774–787. https://doi.org/10.1111/ecog.02671
Dinerstein E, Olson D, Joshi A, Vynne C, Burgess ND, Wikramanayake E, Hann N, Palminteri S, Hedao P, Noss R, Hansen M, Locke H, Ellis HC, Jones B, Barber CV, Hayes R, Kormos C, Martin V, Crist E, Sechrest W, Price L, Baillie JEM, Weeden D, Suckling K, Davis C, Sizer N, Moore R, Thau D, Birch T, Potapov P, Turubanova S, Tyukavina A, de Souza N, Pintea L, Brito JC, Llewellyn OA, Miller AG, Patzelt A, Ghazanfar SA, Timberlake J, Klöser H, Shennan-Farpón Y, Kindt R, Barnekow Lillesø J-P, van Breugel P, Graudal L, Voge M, Al-Shammari KF, Saleem M (2017) An ecoregion-based approach to protecting half the Terrestrial Realm. Bioscience 67:534–545. https://doi.org/10.1093/biosci/bix014
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. https://doi.org/10.1093/molbev/mss075
Durham J, Arellano AV, Peck JH Jr (1955) Evidence for no Cenozoic Isthmus of Tehuantepec seaways. Geol Soc Am Bull 66:977–992. https://doi.org/10.1130/0016-7606(1955)66[977:EFNCIO]2.0.CO;2
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mo Ecol Res 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fay MP, Proschan MA (2010) Wilcoxon-Mann-Whitney or t-test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Stat Surv 4:1–39. https://doi.org/10.1214/09-SS051
Fossøy F, Sorenson MD, Liang W, Ekrem T, Moksnes A, Møller AP, Stokke BG (2016) Ancient origin and maternal inheritance of blue cuckoo eggs. Nat Commun 7:10272. https://doi.org/10.1038/ncomms10272
Fritz U, Hundsdörfer AK, Široký P, Auer M, Kami H, Lehmann J, Wink M (2007) Phenotypic plasticity leads to incongruence between morphology-based taxonomy and genetic differentiation in western Palaearctic tortoises (Testudo graeca complex; Testudines, Testudinidae). Amphibia-Reptilia 28:97–121. https://doi.org/10.1163/156853807779799135
Funk DJ, Omland KE (2003) Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst 34:397–423
García-Moreno J, Navarro-Sigüenza AG, Peterson AT, Sánchez-González LA (2004) Genetic variation coincides with geographic structure in the common bush-tanager (Chlorospingus ophthalmicus) complex from Mexico. Mol Phylogenet Evol 33:186–196. https://doi.org/10.1016/j.ympev.2004.05.007
GBIF.org (2021) GBIF Occurrence Download https://doi.org/10.15468/dl.e3e8vg
Gill FB (2014) Species taxonomy of birds: Which null hypothesis? Auk 131:150–161. https://doi.org/10.1642/AUK-13-206.1
Gill F, Donsker D (eds) (2017) IOC World Bird List (v 7.2). https://doi.org/10.14344/IOC.ML.7.2
Glor RE, Warren D (2011) Testing ecological explanations for biogeographic boundaries. Evolution 65:73–683. https://doi.org/10.1111/j.1558-5646.2010.01177.x
Graham A, Dilcher D (1995) The Cenozoic record of tropical dry forest in northern Latin America and the southern United States. In: Bullock SH, Mooney HA (eds) Seasonally Dry Tropical Forests. Cambridge University Press, Cambridge, pp 124–145
Hackett SJ (1996) Molecular phylogenetics and biogeography of tanagers in the genus Ramphocelus (Aves). Mol Phylogenet Evol 5:368–382. https://doi.org/10.1006/mpev.1996.0032
Hamilton M (2009) Population genetics. John Wiley and Sons, Hoboken
Hasumi H, Emori S (2004) K-1 coupled GCM (MIROC) description. Center for Climate System Research, University of Tokyo, Tokyo
Hedenström A (2008) Adaptations to migration in birds: behavioural strategies, morphology and scaling effects. Philos Trans R Soc Lond B Biol Sci 363:287–299. https://doi.org/10.1098/rstb.2007.2140
Hernández-Canchola G, León-Paniagua L (2017) Genetic and ecological processes promoting early diversification in the lowland Mesoamerican bat Sturnira parvidens (Chiroptera: Phyllostomidae). Mol Phylogenet Evol 114:334–345. https://doi.org/10.1016/j.ympev.2017.06.015
Hernández-Romero PC, Gutiérrez-Rodríguez C, Valdespino C, Prieto-Torres DA (2018) The role of geographical and ecological factors on population divergence of the Neotropical otter Lontra longicaudis (Carnivora, Mustelidae). Evol Biol 45:37–55. https://doi.org/10.1007/s11692-017-9428-5
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Javis A (2005) Very high-resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Holmes RT (1986) Foraging patterns of forest birds: male-female differences. Wilson Bull 98:196–213. http://www.jstor.org/stable/4162226. Accessed 3 Sep 2020
Hosner PA, Sánchez-González LA, Peterson AT, Moyle RG (2014) Climate-driven diversification and pleistocene refugia in Philippine birds: evidence from phylogeographic structure and paleoenvironmental niche modeling. Evolution 68:2658–2674. https://doi.org/10.1111/evo.12459
Howell SNG, Webb S (1995) A guide to the birds of Mexico and Northern Central America. Oxford University Press, New York
Johnson NK, Cicero C (2004) New mitochondrial DNA data affirm the importance of Pleistocene speciation in North American birds. Evolution 58:1122–1130. https://doi.org/10.1111/j.0014-3820.2004.tb00445.x
Johnson KP, Sorenson MD (1998) Comparing molecular evolution in two mitochondrial protein coding genes (cytochrome b and ND2) in the dabbling ducks (Tribe: Anatini). Mol Phylogenet Evol 10:82–94. https://doi.org/10.1006/mpev.1997.0481
Jønsson KA, Lessard JP, Ricklefs RE (2015) The evolution of morphological diversity in continental assemblages of passerine birds. Evolution 69:879–889. https://doi.org/10.1111/evo.12622
Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90:773–795. https://doi.org/10.1080/01621459.1995.10476572
Komar O, Thurber WA (2003) Predation on Birds by a Cuckoo (Cuculidae), Mockingbird (Mimidae), and Saltator (Cardinalidae). Wilson Bull 115:205–208. https://doi.org/10.1676/02-115
Lanfear R, Calcott B, Ho SY, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701. https://doi.org/10.1093/molbev/mss020
Leigh JW, Bryant D (2015) PopArt: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Lerner HR, Meyer M, James HF, Hofreiter M, Fleischer RC (2011) Multilocus resolution of phylogeny and timescale in the extant adaptive radiation of Hawaiian honeycreepers. Curr Biol 21:1838–1844. https://doi.org/10.1016/j.cub.2011.09.039
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Lim HC, Sheldon FH (2011) Multilocus analysis of the evolutionary dynamics of rainforest bird populations in Southeast Asia. Mol Ecol 20:3414–3438. https://doi.org/10.1111/j.1365-294X.2011.05190.x
Liu C, White M, Newell G (2013) Selecting thresholds for the prediction of species occurrence with presence‐only data. J Biogeogr 40:778–789. https://doi.org/10.1111/jbi.12058
Liu C, Newell G, White M (2016) On the selection of thresholds for predicting species occurrence with presence‐only data. Ecol Evol 6: 337-348. https://doi.org/10.1002/ece3.1878
McCormack JE, Peterson AT, Bonaccorso E, Smith TB (2008) Speciation in the highlands of Mexico: genetic and phenotypic divergence in the Mexican jay (Aphelocoma ultramarina). Mol Ecol 17:2505–2521. https://doi.org/10.1111/j.1365-294X.2008.03776.x
Mendonça P, Dias C, Aleixo A, Carneiro LS, Araripe J, Rêgo PSD (2021) Diversification across the Isthmus of Tehuantepec explains the phylogeographic arrangement of the widespread bright-rumped Attila (Attila spadiceus; Tyrannidae) and reveals the existence of two major lineages. J Ornithol. https://doi.org/10.1007/s10336-021-01928-3
Miles DB, Ricklefs RE (1984) The correlation between ecology and morphology in deciduous forest passerine birds. Ecology 65:1629–1640. https://doi.org/10.2307/1939141
Miller KG, Kominz MA, Browning JV, Wright JD, Mountain GS, Katz ME, Sugraman PJ, Cramer BS, Christie-Blick N, Pekar SF (2005) The Phanerozoic record of global sea-level change. Science 310:1293–1298. https://doi.org/10.1126/science.1116412
Molnar P, Cane MA (2007) Early Pliocene (pre-Ice age) El Niño-like global climate: which El Niño? Geosphere 3:337–365. https://doi.org/10.1130/GES00103.1
Montaño-Rendón M, Sánchez-González LA, Hernández-Alonso G, Navarro-Sigüenza AG (2015) Genetic differentiation in the Mexican endemic Rufous-backed Robin, Turdus rufopalliatus (Passeriformes: Turdidae). Zootaxa 4034:495–514. https://doi.org/10.11646/zootaxa.4034.3.4
Moreno-Contreras I, Sánchez-González LA, del Coro AM, Prieto-Torres DA, Navarro-Sigüenza AG (2020) Climatic niche evolution in the Arremon brunneinucha complex (Aves: Passerellidae) in a Mesoamerican landscape. Evol Biol 47:1–10. https://doi.org/10.1007/s11692-020-09498-7
Moritz C, Patton JL, Schneider CJ, Smith TB (2000) Diversification of rainforest faunas: an integrated molecular approach. Annu Rev Ecol Evol Syst 31:533–563. https://doi.org/10.1146/annurev.ecolsys.31.1.533
Morrone JJ (2001) Biogeografía de América Latina y el Caribe, vol 3. M and T-Manuales and Tesis SEA, Zaragoza
Morrone JJ (2014) Biogeographical regionalisation of the Neotropical region. Zootaxa 3782:1–110. https://doi.org/10.11646/zootaxa.3782.1.1
Morrone JJ (2017) Neotropical biogeography: regionalization and evolution. CRC Press, Boca Raton
Mota-Vargas C, Rojas-Soto OR (2016) Taxonomy and ecological niche modeling: implications for the conservation of wood partridges (genus Dendrortyx). J Nat Conserv 29:1–13. https://doi.org/10.1016/j.jnc.2015.10.003
Mulcahy DG, Morrill BH, Mendelson JR III (2006) Historical biogeography of lowland species of toads (Bufo) across the Trans-Mexican Neovolcanic Belt and the Isthmus of Tehuantepec. J Biogeogr 33:1889–1904. https://doi.org/10.1111/j.1365-2699.2006.01546.x
Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. https://doi.org/10.1038/35002501
Naimi B (2015) usdm: Uncertainty analysis for species distribution models. R package 595 version 1, 596. https://cran.r-project.org/web/packages/usdm/. Accessed on 3 Sep 2020
Navarro-Sigüenza AG, Peterson AT (2004) An alternative species taxonomy of the birds of Mexico. Biota Neotrop 4:1–32. https://doi.org/10.1590/S1676-06032004000200013
Navarro-Sigüenza AG, Peterson AT, Nyari A, García-Deras GM, García-Moreno J (2008) Phylogeography of the Buarremon brush-finch complex (Aves, Emberizidae) in Mesoamerica. Mol Phylogenet Evol 47:21–35. https://doi.org/10.1016/j.ympev.2007.11.030
Navarro-Sigüenza AG, Rebón-Gallardo M, Gordillo-Martínez A, Peterson AT, Berlanga-García H, Sánchez-González LA (2014) Biodiversidad de aves en México. Rev Mex Biodivers 85:S476–S495. https://doi.org/10.7550/rmb.41882
Nichols R (2002) Gene trees and species trees are not the same. Trends Ecol Evol 16:358–364
Ornelas JF, Sosa V, Soltis DE, Daza JM, González C, Soltis PS, Gutiérrez-Rodríguez C, de los Monteros EA, Castoe TA, Bell C, Ruiz-Sánchez E (2013) Comparative phylogeographic analyses illustrate the complex evolutionary history of threatened cloud forests of northern Mesoamerica. PLoS ONE 8:e56283. https://doi.org/10.1371/journal.pone.0056283
Ortiz-Ramírez MF, Andersen MJ, Zaldívar-Riverón A, Ornelas JF, Navarro-Sigüenza AG (2016) Geographic isolation drives divergence of uncorrelated genetic and song variation in the Ruddy-capped Nightingale-Thrush (Catharus frantzii; Aves: Turdidae). Mol Phylogenet Evol 94:74–86. https://doi.org/10.1016/j.ympev.2015.08.017
Ottvall R, Bensch S, Walinder G, Lifjeld JT (2002) No evidence of genetic differentiation between lesser redpolls Carduelis flammea cabaret and common redpolls Carduelis f. flammea. Avian Science 2:237–244
Owens HL, Campbell LP, Dornak LL, Saupe EE, Barve N, Soberón J, Ingenloff K, Lira-Noriega A, Hensz CM, Myers CE, Peterson AT (2013) Constraints on interpretation of ecological niche models by limited environmental ranges on calibration areas. Ecol Modell 263:10–18. https://doi.org/10.1016/j.ecolmodel.2013.04.011
Paterlini M (2007) There shall be order. the legacy of Linnaeus in the age of molecular biology. EMBO Rep 8:814–816. https://doi.org/10.1038/sj.embor.7401061
Payne RB (2005) The cuckoos. Oxford University Press, New York
Payne RB (1997) Cuculiformes. In:del Hoyo J, Elliott A, Sargatal J (eds). Handbook of the Birds of the World vol. 4. Sandgrouse to Cuckoos. Lynx Edicions, Barcelona
Pennington RT, Prado DE, Pendry CA (2000) Neotropical seasonally dry forest and quaternary vegetation changes. J Biogeogr 27:261–273. https://doi.org/10.1046/j.1365-2699.2000.00397.x
Peters JL (1940) Check-list of birds of the world, vol 4. Museum of comparative zoology, Cambridge
Peterson AT, Navarro AG (2000) Western Mexico: a significant centre of avian endemism and challenge for conservation action. Cotinga 14:42–46
Peterson AT, Soberón J, Sánchez-Cordero V (1999) Conservatism of ecological niches in evolutionary time. Science 285:1265–1267. https://doi.org/10.1126/science.285.5431.1265
Peterson AT, Papeş M, Soberón J (2008) Rethinking receiver operating characteristic analysis applications in ecological niche modeling. Ecol Modell 213:63–72. https://doi.org/10.1016/j.ecolmodel.2007.11.008
Peterson AT, Soberón J, Pearson RG, Anderson RP, Martínez-Meyer E, Nakamura M, Araújo MB (2011) Ecological niches and geographic distributions. Princeton University Press, New Jersey
Phillips AR (1986) The known birds of North and Middle America, Part 1. In: Phillips AR (ed) Hirundinidae to Mimidae; Certhiidae. Denver Museum of Natural History, Denver
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Modell 190:231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026
Piratelli A, Pereira MR (2002) Dieta de aves na região leste de Mato Grosso do Sul, Brasil. Ararajuba 10:131–139
Posada D, Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16:37–45. https://doi.org/10.1016/S0169-5347(00)02026-7
Prieto-Torres DA, Rojas-Soto OR, Bonaccorso E, Santiago-Alarcón D, Navarro-Sigüenza AG (2019a) Distributional patterns of Neotropical seasonally dry forest birds: a biogeographical regionalization. Cladistics 35:446–460. https://doi.org/10.1111/cla.12366
Prieto-Torres DA, Rojas-Soto OR, Santiago-Alarcón D, Bonaccorso E, Navarro-Sigüenza AG (2019b) Diversity, endemism, species turnover and relationships among avifauna of Neotropical seasonally dry forests. Ardeola 66:257–277. https://doi.org/10.13157/arla.66.2.2019.ra1
Prum RO, Berv JS, Dornburg A, Field DJ, Townsend JP, Lemmon EM, Lemmon AR (2015) A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 526:569–573. https://doi.org/10.1038/nature15697
Puebla-Olivares F, Bonaccorso E, De Los Monteros AE, Omland KE, Llorente-Bousquets JE, Peterson AT, Navarro-Sigüenza AG (2008) Speciation in the emerald toucanet (Aulacorhynchus prasinus) complex. Auk 125:39–50. https://doi.org/10.1525/auk.2008.125.1.39
R Core Team (2014) R: A Language and environment for statistical computing. R Foundation for statistical computing, Vienna. http://www.R-project.org/. Accessed 3 Sep 2020
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer. Accessed 3 Sep 2020
Ramírez-Barrera M, Hernández-Baños BE, Jaramillo JP, Klicka J (2018) Deep divergence of red-crowned Ant Tanager (Habia rubica: Cardinalidae), a multilocus phylogenetic analysis with emphasis in Mesoamerica. PeerJ 6:e5496. https://doi.org/10.7717/peerj.5496
Raw A (1997) Avian predation on individual neotropical social wasps (Hymenoptera, Vespidae) outside their nests. Ornitol Neotrop 8:89–92
Repenning M, Basso HCDP, Rossoni JR, Krügel MM, Fontana CS (2009) Análise comparativa da dieta de quatro espécies de cucos (Aves: Cuculidae), no sul do Brasil. Zoologia 26:443–453. https://doi.org/10.1590/S1984-46702009000300008
Reyes-Velasco J, Meik JM, Smith EN, Castoe TA (2013) Phylogenetic relationships of the enigmatic longtailed rattlesnakes (Crotalus ericsmithi, C. lannomi, and C. stejnegeri). Mol Phylogenet Evol 69:524–534. https://doi.org/10.1016/j.ympev.2013.07.025
Ridgway R (1916) The birds of North and Middle America: a descriptive catalogue of the higher groups, genera, species, and subspecies of birds known to occur in North America, from the Arctic lands to the Isthmus of Panama, the West Indies and other islands of the Caribbean Sea, and the Galapagos Archipelago. United States National Museum Smithsonian Institution, Washington
Ríos-Muñoz CA, Navarro-Sigüenza AG (2012) Patterns of species richness and biogeographic regionalization of the avifaunas of the seasonally dry tropical forest in Mesoamerica. Stud Neotrop Fauna Environ 47:171–182. https://doi.org/10.1080/01650521.2012.734175
Rising JD, Somers KM (1989) The measurement of overall body size in birds. Auk 106:666–674. https://doi.org/10.1093/auk/106.4.666
Rocha-Méndez A, Sánchez-González LA, Arbeláez-Cortés E, Navarro-Sigüenza AG (2018) Phylogeography indicates incomplete genetic divergence among phenotypically differentiated montane forest populations of Atlapetes albinucha (Aves, Passerellidae). ZooKeys 809:125. https://doi.org/10.3897/zookeys.809.28743
Rocha-Méndez A, Sánchez-González LA, González C, Navarro-Sigüenza AG (2019) The geography of evolutionary divergence in the highly endemic avifauna from the Sierra Madre del Sur, Mexico. BMC Evol Biol 19:1–21
Rodríguez-Gómez F, Gutiérrez-Rodríguez C, Ornelas JF (2013) Genetic, phenotypic and ecological divergence with gene flow at the Isthmus of Tehuantepec: the case of the azure-crowned hummingbird (Amazilia cyanocephala). J Biogeogr 40:1360–1373. https://doi.org/10.1111/jbi.12093
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. https://doi.org/10.1093/bioinformatics/btg180
Sánchez-González LA, Moyle RG (2011) Molecular systematics and species limits in the Philippine fantails (Aves: Rhipidura). Mol Phylogenet Evol 61:290–299. https://doi.org/10.1016/j.ympev.2011.06.013
Sánchez-González LA, Hosner PA, Moyle RG (2015) Genetic differentiation in insular lowland rainforests: insights from historical demographic patterns in Philippine Birds. PLoS ONE 10(8):e0134284. https://doi.org/10.1371/journal.pone.0134284
Sánchez-González LA, Castillo-Chora VJ, Arbeláez-Cortés E, Navarro-Sigüenza AG (2021) Diversification and secondary contact in the magpie-jays (Calocitta) throughout the pacific lowlands of Mesoamerica. J Zool Syst Evol Res 59:2371–2386. https://doi.org/10.1111/jzs.12571
Sarkar S, Sánchez-Cordero V, Londoño MC, Fuller T (2009) Systematic conservation assessment for the Mesoamerica, chocó, and tropical andes biodiversity hotspots: a preliminary analysis. Biodivers Conserv 18:1793–1828. https://doi.org/10.1007/s10531-008-9559-1
Sharpe RB (1900) Hand-list of the genera and species of birds, vol 2. British Museum, London
Smith BT, Klicka J (2010) The profound influence of the late pliocene Panamanian uplift on the exchange, diversification, and distribution of new world birds. Ecography 33:333–342. https://doi.org/10.1111/j.1600-0587.2009.06335.x
Smith BT, McCormack JE, Cuervo AM, Hickerson MJ, Aleixo A, Cadena CD, Pérez-Emán J, Burney CW, Xie X, Harvey MG, Faircloth BC, Glenn TC, Derryberry EP, Prejean S, Fields S, Brumfield RT (2014) The drivers of tropical speciation. Nature 515:406–409. https://doi.org/10.1038/nature13687
Soberon J, Peterson AT (2005) Interpretation of models of fundamental ecological niches and species’ distributional areas. Biodiv Inf 2:1–10. https://doi.org/10.17161/bi.v2i0.4
Sorenson MD, Payne RB (2005) The cuckoos. Oxford University Press, New York
Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP (1999) Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogenet Evol 12:105–114. https://doi.org/10.1006/mpev.1998.0602
Stacklies W, Redestig H, Scholz M, Walther D, Selbig J (2007) pcaMethods—a bioconductor package providing PCA methods for incomplete data. Bioinformatics 23:1164–1167. https://doi.org/10.1093/bioinformatics/btm069
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. https://doi.org/10.1093/bioinformatics/btl446
Stone W (1908) A review of the genus Piaya lesson. Proc Acad Nat Sci Phila 60:492–501
Sullivan J, Arellano E, Rogers DS (2000) Comparative phylogeography of Mesoamerican highland rodents: concerted versus independent response to past climatic fluctuations. Am Nat 155:755–768
Svenning JC, Skov F (2004) Limited filling of the potential range in European tree species. Ecol Lett 7:565–573. https://doi.org/10.1111/j.1461-0248.2004.00614.x
Terribile LC, Lima-Ribeiro MS, Araújo MB, Bizão N, Collevatti R, Dobrovolski R, Franco A, Guilhaumon F, Lima J, Murakami DM, Nabout JC, Oliveira G, Oliveira LK, Rabelo SG, Rangel TF, Simon LM, Soares TN, Telles MPC, Diniz-Filho JAF (2012) Areas of climate stability of species ranges in the Brazilian Cerrado: disentangling uncertainties through time. Nat Conserv 10:152–159
Vázquez-López M, Morrone JJ, Ramírez-Barrera SM, López-López A, Robles-Bello SM, Hernández-Baños BE (2020) Multilocus, phenotypic, behavioral, and ecological niche analyses provide evidence for two species within Euphonia affinis (Aves, Fringillidae). ZooKeys 952:129. https://doi.org/10.3897/zookeys.952.51785
Vázquez-Miranda H, Navarro-Sigüenza AG, Omland KE (2009) Phylogeography of the Rufous-naped Wren (Campylorhynchus rufinucha): speciation and hybridization in Mesoamerica. Auk 126:765–778. https://doi.org/10.1525/auk.2009.07048
Voelker G, Outlaw RK, Bowie RC (2010) Pliocene forest dynamics as a primary driver of African bird speciation. Glob Ecol Biogeogr 19:111–121. https://doi.org/10.1111/j.1466-8238.2009.00500.x
Warren DL, Glor RE, Turelli M (2008) Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62:2868–2883. https://doi.org/10.1111/j.1558-5646.2008.00482.x
Warren DL, Matzke NJ, Cardillo M, Baumgartner JB, Beaumont LJ, Turelli M, Glor RE, Huron NA, Simões M, Iglesias TL, Piquet JC, Dinnage R (2021) ENMTools 10: an R package for comparative ecological biogeography. Ecography. https://doi.org/10.1111/ecog.05485
Wei T, Simko V (2017) R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). https://github.com/taiyun/corrplot. Accessed on 3 Sep. 2020.
Weir JT (2006) Divergent timing and patterns of species accumulation in lowland and highland neotropical birds. Evolution 60:842–855. https://doi.org/10.1111/j.0014-3820.2006.tb01161.x
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
Whitmore FC, Stewart RH (1965) Miocene mammals and central american seaways: fauna of the canal zone indicates separation of central and south america during most of the tertiary. Science 148:180–185. https://doi.org/10.1126/science.148.3667.180
Wiens JJ (2004) Speciation and ecology revisited: phylogenetic niche conservatism and the origin of species. Evolution 58:193–197. https://doi.org/10.1111/j.0014-3820.2004.tb01586.x
Wiens JJ, Graham CH (2005) Niche conservatism: integrating evolution, ecology, and conservation biology. Annu Rev Ecol Evol Syst 36:519–539. https://doi.org/10.1146/annurev.ecolsys.36.102803.095431
Xie W, Lewis PO, Fan Y, Kuo L, Chen MH (2011) Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst Biol 60:150–160. https://doi.org/10.1093/sysbio/syq085
Yezerinac SM, Lougheed SC, Handford P (1992) Measurement error and morphometric studies: statistical power and observer experience. Syst Biol 41:471–482. https://doi.org/10.1093/sysbio/41.4.471
Zink RM, Barrowclough GF (2008) Mitochondrial DNA under siege in avian phylogeography. Molecular Ecol 17:2107–2121
Zink RM, Remsen JV Jr (1986) Evolutionary processes and patterns of geographic variation in birds. In: Johnston F (ed) Current Ornithology, vol 4. Plenum Press, New York, pp 1–6
Zink RM, Blackwell-Rago RC, Ronquist F (2000) The shifting roles of dispersal and vicariance in biogeography. Proc R Soc Lond B Biol Sci 267:497–503. https://doi.org/10.1098/rspb.2000.1028
Acknowledgements
We sincerely thank Ella Vázquez-Domínguez, Erick A. García-Trejo, and Alejandro Zaldívar Riverón for comments on earlier drafts of the manuscript. This project was supported by PAPIIT grants (IA-202813 and IN-222817) to LASG. A special thanks to Lynna Kiere, who commented and reviewed the English in our manuscript. We thank the University of Kansas (M. B. Robbins and A. T. Peterson), the Academy of Sciences of Drexel (Nate Rice and Jason Weckstein), and the Museo de Zoología de la Facultad de Ciencias “Alfonso L. Herrera” for kindly providing tissue samples used in this study. We would also like to recognize all the scientific collectors who obtained and deposited samples and voucher specimens, as well as to the local communities for access to lands under their care. We appreciate comments by two anonymous reviewers, the Associate Editor and Franz Bairlein. The present paper is part of the Master’s thesis of Héctor Cayetano, who received a scholarchip from CONACyT (No. 330550).
Funding
This research was funded by PAPIIT-UNAM through projects IA-202813 and IN-222817 to LAS-G.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethics approval
Specimens and tissues used in this project were collected under permits granted by the Secretaría del Medio Ambiente y Recursos Naturales (SEMARNAT) of the Mexican Government.
Consent to participate
All authors agreed to participate in this manuscript.
Consent for publication
All authors read and approved results in this manuscript.
Additional declarations for articles in life science journals that report the results of studies involving humans and/or animals
Not applicable.
Additional information
Communicated by A. Aleixo.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10336_2022_2008_MOESM1_ESM.docx
Supplementary file1 (DOCX 489 KB) SF 1 Methods and procedures used to perform ENM) to define the historical potential distribution areas under Quaternary and Neogene scenarios for each Mexican lineage, including the results from the mobility-oriented parity (MOP) metrics.
10336_2022_2008_MOESM2_ESM.xlsx
Supplementary file2 (XLSX 49 KB) SF II Genbank sequences numbers (to be provided upon acceptance), individual used for morphometric analyses, and distribution localities for the Ecological Niche Modelling (ENM) analyses
10336_2022_2008_MOESM3_ESM.docx
Supplementary file3 (DOCX 20 KB) Table S1 Substitution models and partitions used for the estimation of the phylogenetic trees. Table S2 Population genetics parameters for Piaya cayana. Haplotypes (h); Haplotype diversity (Hd); Nucleotide diversity (π), Historical effective population size (Ne). Table S3 Morphometric variables correlation matrix. BL=Bill length, BW= Bill width, BD= Bill deep, WC= Wing chord, TARSUS= Tarsus length, HALLUX= Hallux length, TAIL= Tail length, and TIPS= length of the white tips on the tail. Table S4 Eigenvectors of the first three principal components and explained variance for each component. Abbreviations as in Table S3.
10336_2022_2008_MOESM4_ESM.pdf
Supplementary file4 (PDF 534 KB) SFig. 1 Phylogenetic tree based on a comprhensive Cytb sampling for the South American group. Numbers above the branches correspond to Bayesian posterior probabilities. Note the non-significant support for the relationship between P. mexicana and the single sequence available for P. c. melanocrissa from the Ecuadorian Chocó.
10336_2022_2008_MOESM5_ESM.pdf
Supplementary file5 (PDF 161 KB) SFig. 2 Haplotype network based on partial Cytb (622bp) sequences. The Chocoan P. c. nigricrissa is placed in a Trans-Andean clade including P. mexicana and P. thermophila.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sánchez-González, L.A., Cayetano, H., Prieto-Torres, D.A. et al. The role of ecological and geographical drivers of lineage diversification in the Squirrel cuckoo Piaya cayana in Mexico: a mitochondrial DNA perspective. J Ornithol 164, 37–53 (2023). https://doi.org/10.1007/s10336-022-02008-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10336-022-02008-w