
Abstract
Nonlesional focal epilepsies (nlFE) represent a heterogenous group of syndromes. They encompass self-limited focal epilepsies of childhood and youth, rare focal, familial epilepsies, epilepsies associated with brain somatic variants, and to a large extent nonfamilial epilepsies that have a complex genetic or unknown background. Genetic testing should be performed in cases of a family history suggestive of monogenic inheritance and in cases that show additional symptoms, such as intellectual impairment, autism, or dysmorphic features. Whole-exome or whole-genome sequencing is the method of choice. Growing evidence suggests including genetic testing also in the presurgical workup of individuals with drug-resistant epilepsy. While individuals that harbor variants in genes of the mammalian target of rapamycin (mTOR) pathway tend to achieve better seizure control following epilepsy surgery, the postsurgical outcome of genetic epilepsies associated with channel function or synaptic transmission appears to be poor. The aim of this article is to review the genetic background of focal epilepsies that occur or persist in adults, provide guidance for genetic testing, and discuss potential implications for presurgical evaluation.
Zusammenfassung
Nichtläsionelle fokale Epilepsien (nlFE) stellen eine heterogene Gruppe von Syndromen dar. Sie umfassen selbstlimitierende Epilepsien des Kindes- und Jugendalters, seltene familiäre fokale Epilepsiesyndrome, durch auf das Gehirn beschränkte somatische Mosaike ausgelöste Epilepsien sowie zu einem großen Anteil nichtfamiliäre nlFE, welche einen komplexen genetischen oder unbekannten Hintergrund aufweisen. Eine genetische Diagnostik sollte in Fällen mit positiver, auf eine monogene Vererbung hindeutende Familienanamnese erfolgen. Überdies sollte eine genetische Testung bei Vorliegen zusätzlicher Symptome wie z. B. Intelligenzminderung, Autismus oder Dysmorphiezeichen angeboten werden. Eine Exom- oder Genomsequenzierung ist die Untersuchungsmethode der Wahl. Es besteht zudem eine wachsende Evidenz, dass eine genetische Testung in die prächirurgische Evaluation pharmakoresistenter Personen integriert werden sollte. So scheinen Individuen mit genetischen Veränderungen in Genen der mammalian target of rapamycin (mTOR)-Signalkaskade von epilepsiechirurgischen Eingriffen zu profitieren, während das postoperative Ergebnis bei genetischen Epilepsien, denen Defekte der Kanalfunktionen oder der synaptischen Transmission zugrunde liegen, oftmals nicht zufriedenstellend ist. Das Ziel dieses Artikels ist die Darstellung des genetischen Hintergrunds fokaler Epilepsiesyndrome, welche bei erwachsenen Personen auftreten bzw. fortbestehen, sowie die Darreichung von Handlungsempfehlungen zur genetischen Testung und prächirurgischen Evaluation.
Similar content being viewed by others


Introduction
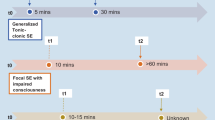
Nonlesional focal epilepsies (nlFE) are characterized by focal seizures, focal interictal epileptic discharges (IEDs), and the absence of epileptogenic lesions on magnetic resonance imaging (MRI). Sometimes nlFE are also described as nonacquired focal epilepsies (NAFE) as opposed to acquired, structural epilepsies, e.g., after cerebral ischemia, hemorrhage, or trauma. Nonlesional focal epilepsies account for 20–40% of all epilepsies [22] and they encompass a wide range of epilepsy syndromes, ranging from self-limited epilepsies in neonates and infants associated with distinct epilepsy genes (e.g., self-limited neonatal epilepsy, SeLNE), self-limited focal epilepsies with presumed complex inheritance in older children (e.g., self-limited epilepsy with centrotemporal spikes, SeLECTS or formerly Rolandic epilepsy), to defined genetic syndromes that begin at a variable age (e.g., epilepsy with auditory features, EAF); see Fig. 1. Yet, the majority of nlFE are isolated and account for a large share of patients seen in daily practice. More than 50% of focal epilepsies do not show structural abnormalities on routine magnetic resonance imaging (MRI; [8]). They can be at best classified by their seizure onset zone while their etiology often remains opaque. In this review, we focus on nlFE that occur or persist in adulthood. For self-limited focal epilepsies of infancy and childhood, overview articles can be found elsewhere [36, 49].

Depiction of syndromic and genetic spectrum of nonlesional focal epilepsies. EAF epilepsy with auditory features, FEs focal epilepsies, MCD malformation of cortical development, FFEVF familial focal epilepsy with variable foci, FMTLE familial mesial temporal lobe epilepsy, POLE photosensitive occipital lobe epilepsy, SeLECTS self-limited epilepsy with centrotemporal spikes, SeLNE self-limited neonatal epilepsy, SHE sleep-related hypermotor epilepsy
With the advent of next-generation sequencing and advances in bioinformatics and statistical genetics, the genetic basis of nlFE was progressively unearthed. Monogenic forms that can be explained by a single, causative gene and feature classic Mendelian pedigrees are the exception. Brain somatic gene variants, i.e., variants present only in local brain tissue, have been found in MRI-negative cases in resective specimens from epilepsy surgery and are associated with microscopic changes of cortical development. The larger share of nlFE, however, seems to rely on the interplay of multiple genetic variants in analogy to idiopathic generalized epilepsies (IGE) and other complex genetic diseases (e.g., schizophrenia or type 2 diabetes; [7, 10]). Yet unlike IGE, which display a more homogeneous phenotype and are firmly established as a polygenic disorder, etiologies in nlFE are presumably more heterogeneous and stretch beyond genetics. Unrecognized autoimmune inflammation, microscopic structural anomalies, and neurodegenerative changes in older individuals offer potential explanations. A positive family history or the presence of certain comorbidities should prompt genetic testing. The indication and consequences of genetic testing in the setting of presurgical evaluation are currently debated but viewed overall favorably.
Nonlesional focal familial epilepsies
Interestingly, while the advancements in DNA sequencing technologies heralded an era of gene discoveries for developmental and epileptic encephalopathies (DEEs), the first epilepsy gene to be identified was CHRNA4. It was detected in a large family with sleep-related frontal lobe seizures [37]. Nonetheless, familial focal epilepsy syndromes are rare. They usually show autosomal-dominant inheritance. But family history may be sparse or not available, and reduced penetrance can further complicate the recognition of a positive family history. Furthermore, seizures in other family members might have been overlooked (e.g., in the case of exclusively nocturnal seizures) or misinterpreted (e.g., in the case of focal aware seizures such as déjà vu auras). Therefore, seizure semiologies associated with familial epilepsies, such as nocturnal hypermotor seizures or focal aware seizures with auditory features, should give rise to a thorough review of the family history [30]. In most cases, affected individuals have normal intellect, normal neurological examination results, and seizures can usually be controlled well [23].
The most current classification differentiates four genetic focal epilepsy syndromes; each can be related to genetic variation in several genes ([30]; see also Table 1).
Sleep-related hypermotor epilepsy
Sleep-related hypermotor epilepsy (SHE), previously also known as “autosomal dominant nocturnal frontal lobe epilepsy” (ADNFLE), usually occurs during adolescence and is characterized by brief nocturnal focal seizures with hyperkinetic, tonic, and dystonic motor features [40], which often appear in clusters after falling asleep or before awakening [33]. Remarkably, some individuals retain consciousness during seizures. Misdiagnoses as parasomnia or dissociative seizures are frequent. The severity among affected family members can show considerable differences and penetrance of the autosomal-dominant disorder is ~ 70% [33], which can be a challenge in smaller families.
Familial mesial temporal lobe epilepsy
Familial mesial temporal lobe epilepsy (FMTLE) usually occurs during youth or early adulthood, notably in individuals without prior febrile seizures. Patients experience nearly exclusively focal aware seizures with pronounced déjà vu, and less commonly other emblematic temporal lobe semiologic features, such as epigastric sensations and anxiety. Focal impaired awareness seizures and bilateral tonic–clonic seizures are rare [24]. Since affected individuals often deem their seizures to be not pathologic, the syndrome is probably underrecognized A systematic study of patients with nonlesional temporal lobe epilepsy identified FMTLE in ~ 20% [24]. Family history might require interviewing family members in person.
Epilepsy with auditory features
Epilepsy with auditory features (EAF) usually has its onset in adolescence or young adulthood [18]. Focal aware seizures with a prominent auditory component or receptive aphasia are the predominant seizure type. Auditory symptoms comprise rather elementary sensations such as humming or buzzing, whereas more complex auditory hallucinations are uncommon [23]. Focal impaired awareness seizures and bilateral tonic–clonic seizures can occur. The inheritance pattern in EAF is autosomal-dominant.
Familial focal epilepsy with variable foci
Familial focal epilepsy with variable foci (FFEVF) is an autosomal-dominant disorder that features a remarkable intrafamilial variability of seizure semiology. Seizures in different family members often arise from different lobes, while being constant within the same individuals [25].
Genetics of nonlesional focal familial epilepsies
Familial nlFE usually exhibit autosomal-dominant inheritance, although penetrance may vary. Variants in nicotinic acetylcholine receptor subunits have been found in SHE, and variants in LGI1 in EAF have been identified thanks to thorough genetic work-up of extended families [3, 37]. Next-generation sequencing technology put the focus on the gene DEPDC5. Together with NPRL2 and NPRL3 it forms the GATOR1 complex that acts as an inhibitor of the mTOR pathway [1]. The latter mediates essential cellular functions such as cell growth, migration, proliferation, and protein synthesis [16]. Crossing syndromic boundaries, DEPDC5 variants have been found in various forms of familial nlFE such as FFEVF [6], SHE [26], and FMTLE [39]. Variants in DEPDC5 as well as NPRL2 and NPRL3 have also been described in familial focal epilepsies, in which some of the affected individuals displayed focal cortical dysplasia (FCD) whereas others had nlFE [44].
Focal epilepsies related to brain somatic variants
Malformations of cortical development (MCD) that range from regional disturbances of cortical architecture such as FCD to complex, pan-cerebral lesions such as hemimegalencephaly have been shown to be founded on genetic factors [12]. For FCD, however, germline variants appear to play a minor role [2, 34]. Somatic variants in various genes of the mTOR pathway (DEPDC5, MTOR, NPRL2/3) were identified in postsurgical resection tissue in structural lesions [2, 19, 44]. Also, more extended lesions harbor somatic variants in mTOR pathway genes, such as MTOR itself, AKT, and PIK3CA [13]. Of late, with SLC35A2, a “non-MTOR” gene was shown to be related to malformations of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE), a milder form of MCD [4]. Moreover, for various mTOR pathway genes, such as TSC1/TSC2 (causing tuberous sclerosis) and DEPDC5, a double-hit hypothesis has been postulated. Here, the combined effect of a germline variant in conjunction with brain somatic variants is thought to give rise to circumscribed MCD [28, 29]. Unlike germline variants, brain somatic variants arise during cortical development resulting in somatic mosaicism. Depending on the time of occurrence, variants can be limited to small fractions of brain cells. By definition, somatic variants are not detectable in leukocyte-derived DNA samples.
The observation that often FCD are not recognizable in presurgical MRI [27] gives rise to the question of whether a share of nlFE can also be explained by somatic variants in “hidden” FCD. In a series of nlFE patients who underwent epilepsy surgery, somatic variants in SLC35A2 were found in 17% of cases [45]. Interestingly, in two of the three reported patients, the pathologic work-up revealed FCD1a. Ongoing research will probably uncover further genes associated with brain somatic variants and nlFE [9]. Developments in structural MRI and postprocessing techniques will help identify more locally confined lesions in the future [41] and thereby render a share of today’s MRI-negative epilepsies “MRI-positive.”
Nonlesional nonfamilial focal epilepsies
In analogy to IGE, nlFE have been the focus of international, large-scale sequencing and genotyping consortia. Like IGE, nlFE have been shown to rely on complex genetic architecture, albeit to a lesser degree. The role of common genetic variants, i.e., variants that occur with a minor allele frequency of > 1% in the population, was established through genome-wide association studies (GWAS; [10]). These GWAS assess the association of single-nucleotide polymorphisms (SNPs) with complex traits. Since the effect size of each SNP by itself is very low, large cohorts are needed to capture significant associations. The latest and to date largest GWAS for epilepsies, including more than 29,000 individuals with epilepsy [11], showed an SNP-based heritability for nlFE of approximately 16%. In comparison to IGE, which features an SNP-based heritability for nlFE of approximately 40%, this effect appears to be rather moderate. Moreover, while the study identified 19 genome-wide significant loci for IGE, none was identified for nlFE. These data suggest a minor role of common variants for nlFE but could also mirror the higher heterogeneity of the nlFE cohort, potentially including mislabeled cases of acquired focal epilepsy, cases with brain somatic mutations, or unrecognized cases of autoimmune epilepsy that could have diluted the effect. Polygenic risk score (PRS) analyses, which estimate the aggregated effect of all SNPs weighted by their effect size on an individual level, underlined that common variants convey an increased risk for nlFE that is, however, smaller than in IGE [17, 21]. Large-scale analyses of exome sequencing data demonstrated that also ultra-rare missense variants (URVs) and truncating variants were enriched in nlFE [7], albeit less than in IGE. Truncating variants in genes, associated with familial nlFE, such as DEPDC5, appear to be enriched in nonfamilial nlFE [46]. Interestingly, enrichment patterns of URVs seem to differ between nlFE and IGE: While IGE exhibited a higher burden of URVs in gene sets derived from inhibitory neurons, nlFE carried a higher burden of URVs derived from excitatory neurons [14].
Genetic testing in nonlesional focal epilepsies
Genetic testing has become an established component of the diagnostic work-up of individuals with epilepsy. Decreasing costs for genetic testing and broader financial coverage by insurance companies made testing more widely available. The ascertainment of a genetic diagnosis enables genetic counseling, may give diagnostic and therapeutic guidance, and can help avoid further, potentially detrimental diagnostic tests [43]. Genetic testing should be considered for individuals with a high pretest probability. In the case of nlFE, testing should be performed of individuals with a positive family history of epilepsy, especially if the symptoms point to a specific, familial nlFE syndrome, such as SHE or EAF. Since families are often small, family members may not be contactable, and penetrance is usually incomplete, specific attention should be directed to seizure semiologies that are suggestive of familial nlFE syndromes, such as nocturnal hypermotor seizures or auditory, focal aware seizures. In nonfamilial nlFE, genetic testing should be considered in individuals who present with additional symptoms such as intellectual impairment, autism spectrum disorders, or dysmorphic features [15]. In patients with epilepsy and intellectual impairment (IQ < 70), the diagnostic yield of genetic testing can be up to 50% [47]. In this cohort, besides missense variants, also copy number variants, i.e., deletions or duplications of large DNA segments, account for about one third of positive cases.
If genetic testing is performed, either whole-exome sequencing (WES) or whole-genome sequencing (WGS) should be favored. Epilepsy gene panels that contain a curated list of known epilepsy-associated genes are obsolete since WES and WGS deliver a higher diagnostic yield (up to 45/48% for WES/WGS vs. 25% for panel diagnostics; [31, 35]). If available, WES/WGS should be performed as a trio analysis, i.e., including both parents for the sake of interpretation of unknown variants WES is today the standard in most diagnostic laboratories, and analysis pipelines usually include CNV analyses, which render chromosomal microarrays redundant. See Table 2 for an overview of indications for genetic testing in nlFE. General recommendations for genetic testing in individuals with epilepsy in Germany are published and regularly reviewed by the Epilepsy and Genetics Commission (Kommission Epilepsie und Genetik) of the German ILAE branch (DGfE; Dt. Ges. für Epileptologie; http://www.dgfe.org/home/index,id,528.html) and have recently been published by the ILAE genetics commission [15].
Genetic testing in the presurgical evaluation of drug-resistant nlFE
Resective epilepsy surgery for individuals with genetic epilepsies may seem counterintuitive at first glance. Yet, as in tuberous sclerosis, resective surgery has been established for many years [48]. Increasing knowledge about the associations of mTOR-pathway genes beyond TSC1 and TSC2 and their association with focal lesional as well as nonlesional epilepsies has kindled the debate about whether patients with nlFE should systematically undergo genetic testing during presurgical work-up [20, 38]. A better understanding of the relation between genetic diagnosis and postoperative outcome could empower clinicians to make better predictions about the odds of successful epilepsy surgery. Systematic reviews and large case collections testify to the low chances of effective seizure control in patients carrying variants in genes related to channel function or synaptic transmission [5, 38]. The same studies observe far more promising results for variants in mTOR-pathway genes. This trend is corroborated by findings from a Dutch epilepsy center that also highlights the increasing use of genetic testing in nlFE [32]. Although clinicians should be wary of performing surgery in patients with channelopathies or synaptopathies, these individuals should not be categorically excluded from presurgical evaluation [42]. A recent survey by the DGfE among German epilepsy centers showed that genetic testing is viewed favorably in many case constellations and, in the case of nlFE, recommended by more than 90% of the survey respondents [5]. Founded on these results, the Epilepsy and Genetics Commission (DGfE) recommends genetic testing as part of the presurgical work-up.
Practical conclusion
-
Individuals with nonlesional focal epilepsy (nlFE) should undergo a thorough examination for positive family history and hallmarks of specific familial nlFE (e.g., nocturnal hypermotor seizures, auditory seizures).
-
Genetic testing should be performed on individuals with a family history suggestive of monogenic inheritance, patients with defined syndromes (e.g., epilepsy with auditory features), and individuals with additive symptoms (intellectual impairment, autism, dysmorphic features).
-
Genetic testing should be considered during presurgical evaluation of patients with drug-resistant focal epilepsy.
References
Baldassari S, Licchetta L, Tinuper P, Bisulli F, Pippucci T (2016) GATOR1 complex: the common genetic actor in focal epilepsies. J Med Genet 53:503–510
Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, Nordli D, Cossette P, Nguyen S, Lambrecq V, Vlaicu M, Daniau M, Bielle F, Andermann E, Andermann F, Leguern E, Chassoux F, Picard F (2015) Familial focal epilepsy with focal cortical dysplasia due to DEPDC 5 mutations: FCD and DEPDC5 mutations. Ann Neurol 77:675–683
Berkovic SF, Izzillo P, McMahon JM, Harkin LA, McIntosh AM, Phillips HA, Briellmann RS, Wallace RH, Mazarib A, Neufeld MY, Korczyn AD, Scheffer IE, Mulley JC (2004) LGI1 mutations in temporal lobe epilepsies. Neurology 62:1115–1119
Bonduelle T, Hartlieb T, Baldassari S, Sim NS, Kim SH, Kang H‑C, Kobow K, Coras R, Chipaux M, Dorfmüller G, Adle-Biassette H, Aronica E, Lee JH, Blumcke I, Baulac S (2021) Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). Acta Neuropathol Commun 9:3
Boßelmann CM, Antonio-Arce VS, Schulze-Bonhage A, Fauser S, Zacher P, Mayer T, Aparicio J, Albers K, Cloppenborg T, Kunz W, Surges R, Syrbe S, Weber Y, Wolking S (2022) Genetic testing before epilepsy surgery—an exploratory survey and case collection from German epilepsy centers. Seizure 95:4–10
Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, Crompton DE, Hughes JN, Bellows ST, Klein KM, Callenbach PMC, Corbett MA, Gardner AE, Kivity S, Iona X, Regan BM, Weller CM, Crimmins D, O’Brien TJ, Guerrero-López R, Mulley JC, Dubeau F, Licchetta L, Bisulli F, Cossette P, Thomas PQ, Gecz J, Serratosa J, Brouwer OF, Andermann F, Andermann E, van den Maagdenberg AMJM, Pandolfo M, Berkovic SF, Scheffer IE (2013) Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 45:546–551
Epi25 Collaborative (2019) Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am J Hum Genet 105:267–282
Hakami T, McIntosh A, Todaro M, Lui E, Yerra R, Tan KM, French C, Li S, Desmond P, Matkovic Z, O’Brien TJ (2013) MRI-identified pathology in adults with new-onset seizures. Neurology 81:920–927
Iffland PH 2nd, Crino PB (2019) The role of somatic mutational events in the pathogenesis of epilepsy. Curr Opin Neurol 32:191–197
International League Against Epilepsy Consortium on Complex Epilepsies (2018) Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun 9:5269
International League Against Epilepsy Consortium on Complex Epilepsies (2022) Genome-wide meta-analysis of over 29,000 people with epilepsy reveals 26 loci and subtype-specific genetic architecture (medRxiv:2022.2006.2008.22276120)
Jamuar SS, Lam AT, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, Servattalab S, Mehta BK, Topcu M, Amrom D, Andermann E, Dan B, Parrini E, Guerrini R, Scheffer IE, Berkovic SF, Leventer RJ, Shen Y, Wu BL, Barkovich AJ, Sahin M, Chang BS, Bamshad M, Nickerson DA, Shendure J, Poduri A, Yu TW, Walsh CA (2014) Somatic mutations in cerebral cortical malformations. N Engl J Med 371:733–743
Jansen LA, Mirzaa GM, Ishak GE, O’Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Rivière JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, Dobyns WB (2015) PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138:1613–1628
Koko M, Krause R, Sander T, Bobbili DR, Nothnagel M, May P, Lerche H (2021) Distinct gene-set burden patterns underlie common generalized and focal epilepsies. EBioMedicine 72:103588
Krey I, Platzer K, Esterhuizen A, Berkovic SF, Helbig I, Hildebrand MS, Lerche H, Lowenstein D, Møller RS, Poduri A, Sadleir L, Sisodiya SM, Weckhuysen S, Wilmshurst JM, Weber Y, Lemke JR, Berkovic SF, Cross JH, Helbig I, Lerche H, Lowenstein D, Mefford HC, Perucca P, Tan NC, Caglayan H, Helbig K, Singh G, Weber Y, Weckhuysen S (2022) Current practice in diagnostic genetic testing of the epilepsies. Epileptic Disord 24:765–786
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Leu C, Stevelink R, Smith AW, Goleva SB, Kanai M, Ferguson L, Campbell C, Kamatani Y, Okada Y, Sisodiya SM, Cavalleri GL, Koeleman BPC, Lerche H, Jehi L, Davis LK, Najm IM, Palotie A, Daly MJ, Busch RM, Epi25 Consortium, Lal D (2019) Polygenic burden in focal and generalized epilepsies. Brain. https://doi.org/10.1515/medgen-2022-2146
Michelucci R, Poza JJ, Sofia V, De Feo MR, Binelli S, Bisulli F, Scudellaro E, Simionati B, Zimbello R, D’Orsi G, Passarelli D, Avoni P, Avanzini G, Tinuper P, Biondi R, Valle G, Mautner VF, Stephani U, Tassinari CA, Moschonas NK, Siebert R, Lopez de Munain A, Perez-Tur J, Nobile C (2003) Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia 44:1289–1297
Møller RS, Weckhuysen S, Chipaux M, Marsan E, Taly V, Bebin EM, Hiatt SM, Prokop JW, Bowling KM, Mei D, Conti V, De La Grange P, Ferrand-Sorbets S, Dorfmüller G, Lambrecq V, Larsen LHG, Leguern E, Guerrini R, Rubboli G, Cooper GM, Baulac S (2016) Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet 2:e118
Moloney P, Dugan P, Widdess-Walsh P, Devinsky O, Delanty N (2022) Genomics in the presurgical epilepsy evaluation. Epilepsy Res 184:106951. https://doi.org/10.1016/j.eplepsyres.2022.106951
Moreau C, Rébillard R‑M, Wolking S, Michaud J, Tremblay F, Girard A, Bouchard J, Minassian B, Laprise C, Cossette P, Girard SL (2020) Polygenic risk scores of several subtypes of epilepsies in a founder population. Neurol Genet 6:e416
Peljto AL, Barker-Cummings C, Vasoli VM, Leibson CL, Hauser WA, Buchhalter JR, Ottman R (2014) Familial risk of epilepsy: a population-based study. Brain 137:795–805
Perucca P (2018) Genetics of focal epilepsies: what do we know and where are we heading? Epilepsy Curr 18:356–362
Perucca P, Crompton DE, Bellows ST, McIntosh AM, Kalincik T, Newton MR, Vajda FJE, Scheffer IE, Kwan P, O’Brien TJ, Tan KM, Berkovic SF (2017) Familial mesial temporal lobe epilepsy and the borderland of déjà vu. Ann Neurol 82:166–176
Picard F, Baulac S, Kahane P, Hirsch E, Sebastianelli R, Thomas P, Vigevano F, Genton P, Guerrini R, Gericke CA, An I, Rudolf G, Herman A, Brice A, Marescaux C, LeGuern E (2000) Dominant partial epilepsies. A clinical, electrophysiological and genetic study of 19 European families. Brain 123(6):1247–1262
Picard F, Makrythanasis P, Navarro V, Ishida S, de Bellescize J, Ville D, Weckhuysen S, Fosselle E, Suls A, De Jonghe P, Vasselon Raina M, Lesca G, Depienne C, An-Gourfinkel I, Vlaicu M, Baulac M, Mundwiller E, Couarch P, Combi R, Ferini-Strambi L, Gambardella A, Antonarakis SE, Leguern E, Steinlein O, Baulac S (2014) DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 82:2101–2106
Porter BE, Judkins AR, Clancy RR, Duhaime A, Dlugos DJ, Golden JA (2003) Dysplasia: a common finding in intractable pediatric temporal lobe epilepsy. Neurology 61:365–368
Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE, Bouffard P, Kwiatkowski DJ (2010) Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol 20:1096–1105
Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, Muraca G, Roussel D, Navarro V, Leguern E, Miles R, Baulac S (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest 128:2452–2458
Riney K, Bogacz A, Somerville E, Hirsch E, Nabbout R, Scheffer IE, Zuberi SM, Alsaadi T, Jain S, French J, Specchio N, Trinka E, Wiebe S, Auvin S, Cabral-Lim L, Naidoo A, Perucca E, Moshé SL, Wirrell EC, Tinuper P (2022) International league against epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE task force on nosology and definitions. Epilepsia 63:1443–1474
Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A (2019) Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology 92:e418–428
Sanders M, Lemmens CMC, Jansen FE, Brilstra EH, Koeleman BPC, Braun KPJ, Dutch Collaborative Epilepsy Surgery (2019) Implications of genetic diagnostics in epilepsy surgery candidates: a single-center cohort study. Epilepsia Open 4:609–617
Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F (1995) Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain 118(1):61–73
Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, Licchetta L, Provini F, Bisulli F, Vadlamudi L, Gecz J, Connelly A, Tinuper P, Ricos MG, Berkovic SF, Dibbens LM (2014) Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 75:782–787
Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, Mulhern MM, Partack EJ, Poduri A (2022) Genetic testing for the epilepsies: a systematic review. Epilepsia 63:375–387
Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, Guerreiro M, Gwer S, Zuberi SM, Wilmshurst JM, Yozawitz E, Pressler R, Hirsch E, Wiebe S, Cross HJ, Perucca E, Moshé SL, Tinuper P, Auvin S (2022) International league against epilepsy classification and definition of epilepsy syndromes with onset in childhood: position paper by the ILAE task force on nosology and definitions. Epilepsia 63:1398–1442
Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11:201–203
Stevelink R, Sanders MW, Tuinman MP, Brilstra EH, Koeleman BP, Jansen FE, Braun KP (2018) Epilepsy surgery for patients with genetic refractory epilepsy: a systematic review. Epileptic Disord 20:99–115
Striano P, Serioli E, Santulli L, Manna I, Labate A, Dazzo E, Pasini E, Gambardella A, Michelucci R, Striano S, Nobile C (2015) DEPDC5 mutations are not a frequent cause of familial temporal lobe epilepsy. Epilepsia 56:e168–171
Tinuper P, Bisulli F, Cross JH, Hesdorffer D, Kahane P, Nobili L, Provini F, Scheffer IE, Tassi L, Vignatelli L, Bassetti C, Cirignotta F, Derry C, Gambardella A, Guerrini R, Halasz P, Licchetta L, Mahowald M, Manni R, Marini C, Mostacci B, Naldi I, Parrino L, Picard F, Pugliatti M, Ryvlin P, Vigevano F, Zucconi M, Berkovic S, Ottman R (2016) Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 86:1834–1842
van Lanen R, Wiggins CJ, Colon AJ, Backes WH, Jansen JFA, Uher D, Drenthen GS, Roebroeck A, Ivanov D, Poser BA, Hoeberigs MC, van Kuijk SMJ, Hoogland G, Rijkers K, Wagner GL, Beckervordersandforth J, Delev D, Clusmann H, Wolking S, Klinkenberg S, Rouhl RPW, Hofman PAM, Schijns O (2022) Value of ultra-high field MRI in patients with suspected focal epilepsy and negative 3 T MRI (EpiUltraStudy): protocol for a prospective, longitudinal therapeutic study. Neuroradiology 64:753–764
Vezyroglou A, Varadkar S, Bast T, Hirsch E, Strobl K, Harvey AS, Epilepsy Surgery in SCN1A Study Group, Scheffer IE, Sisodiya SM, Cross JH (2020) Focal epilepsy in SCN1A-mutation carrying patients: is there a role for epilepsy surgery? Dev Med Child Neurol 62:1331–1335
Weber YG, Biskup S, Helbig KL, von Spiczak S, Lerche H (2017) The role of genetic testing in epilepsy diagnosis and management. Expert Rev Mol Diagn 17:739–750
Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, Baulac M, Fohlen M, Kallay Zetchi C, Seeck M, de la Grange P, Dermaut B, Meurs A, Thomas P, Chassoux F, Leguern E, Picard F, Baulac S (2016) Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57:994–1003
Winawer MR, Griffin NG, Samanamud J, Baugh EH, Rathakrishnan D, Ramalingam S, Zagzag D, Schevon CA, Dugan P, Hegde M, Sheth SA, McKhann GM, Doyle WK, Grant GA, Porter BE, Mikati MA, Muh CR, Malone CD, Bergin AMR, Peters JM, McBrian DK, Pack AM, Akman CI, LaCoursiere CM, Keever KM, Madsen JR, Yang E, Lidov HGW, Shain C, Allen AS, Canoll PD, Crino PB, Poduri AH, Heinzen EL (2018) Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 83:1133–1146
Wolking S, Moreau C, McCormack M, Krause R, Krenn M, Berkovic S, Cavalleri GL, Delanty N, Depondt C, Johnson MR, Koeleman BPC, Kunz WS, Lerche H, Marson AG, O’Brien TJ, Petrovski S, Sander JW, Sills GJ, Striano P, Zara F, Zimprich F, Sisodiya SM, Girard SL, Cossette P (2021) Assessing the role of rare genetic variants in drug-resistant, non-lesional focal epilepsy. Ann Clin Transl Neurol 8:1376–1387
Zacher P, Mayer T, Brandhoff F, Bartolomaeus T, Le Duc D, Finzel M, Heinze A, Horn S, Klöckner C, Körber G, Hentschel J, Kalita M, Krey I, Nastainczyk-Wulf M, Platzer K, Rebstock J, Popp B, Stiller M, Teichmann AC, Jamra RA, Lemke JR (2021) The genetic landscape of intellectual disability and epilepsy in adults and the elderly: a systematic genetic work-up of 150 individuals. Genet Med 23:1492–1497
Zhang K, Hu WH, Zhang C, Meng FG, Chen N, Zhang JG (2013) Predictors of seizure freedom after surgical management of tuberous sclerosis complex: a systematic review and meta-analysis. Epilepsy Res 105:377–383
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, Pressler R, Auvin S, Samia P, Hirsch E, Galicchio S, Triki C, Snead OC, Wiebe S, Cross JH, Tinuper P, Scheffer IE, Perucca E, Moshé SL, Nabbout R (2022) ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE task force on nosology and definitions. Epilepsia 63:1349–1397
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
R. Karge, C. Knopp, Y. Weber and S. Wolking declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards indicated in each case.
Additional information

Scan QR code & read article online
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karge, R., Knopp, C., Weber, Y. et al. Genetics of nonlesional focal epilepsy in adults and surgical implications. Clin Epileptol 36, 91–97 (2023). https://doi.org/10.1007/s10309-023-00568-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10309-023-00568-2
Keywords
- Genetic epilepsies
- Brain somatic variants
- Monogenic disorders
- Complex genetic disorders
- Genetic testing
- Epilepsy surgery