
Abstract
Aplastic anemia (AA) is a potentially fatal bone marrow failure syndrome characterized by a paucity of hematopoietic stem cells and progenitor cells with varying degrees of cytopenia and fatty infiltration of the bone marrow space. Recent advances in genomics have uncovered a link between somatic mutations and myeloid cancer in AA patients. At present, the impact of these mutations on AA patients remains uncertain. We retrospectively investigated 279 AA patients and 174 patients with myelodysplastic syndromes (MDS) and performed targeted sequencing of 22 genes on their bone marrow cells using next-generation sequencing (NGS). Associations of somatic mutations with prognostic relevance and response to treatment were analyzed. Of 279 AA patients, 25 (9.0%) patients had somatic mutations, and 20 (7.2%) patients had one mutation. The most frequently mutated genes were ASXL1(3.2% of the patients), DNMT3A (1.8%) and TET2 (1.8%). In the MDS group, somatic mutations were detected in 120 of 174 (69.0%) patients, and 81 patients (46.6%) had more than one mutation. The most frequently mutated genes were U2AF1 (24.7% of the patients), ASXL1 (18.4%) and TP53 (13.2%). Compared with MDS patients, AA patients had a significantly lower frequency of somatic mutations and mostly one mutation. Similarly, the median variant allele frequency was lower in AA patients than in MDS patients (6.9% vs. 28.4%). The overall response of 3 and 6 months in the somatic mutation (SM) group was 37.5% and 66.7%, respectively. Moreover, there was no significant difference compared with the no somatic mutation (N-SM) group. During the 2-years follow-up period, four (20%) deaths occurred in the SM group and 40 (18.1%) in the N-SM group, with no significant difference in overall survival and event-free survival between the two groups. Our data indicated that myeloid tumor-associated somatic mutations in AA patients were detected in only a minority of patients by NGS. AA and MDS patients had different gene mutation patterns. The somatic mutations in patients with AA were characterized by lower mutation frequency, mostly one mutation, and lower median allelic burden of mutations than MDS. Somatic mutations were a common finding in the elderly, and the frequency of mutations increases with age. The platelet count affected the treatment response at 3 months, and ferritin level affected the outcome at 6 months, while somatic mutations were not associated with treatment response or long-term survival. However, our cohort of patients with the mutation was small; this result needs to be further confirmed with large patient sample.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acquired aplastic anemia (AA) is the prototype of the bone marrow failure syndrome characterized by hypocellular marrow and pancytopenia. The disease is typically acquired, and the principal mechanism is that activated T cells produce proinflammatory cytokines and proteins that are associated with a lack of immune regulation and recognize and eliminate hematopoietic stem or progenitor cells [1,2,3]. The main treatment methods are transplantation and immunosuppressive therapy (IST). Bone marrow transplantation is curative, and patients may have a better response rate to standard immunosuppressive therapy combined with eltrombopag [2, 4]. Long-term follow-up has shown that about 10–20% of AA patients evolved to myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML), especially who did not achieve a complete response following treatment with immunosuppressive therapy [5, 6].
With the advent of next-generation sequencing (NGS), the presence of acquired somatic mutations in myeloid candidate genes previously associated with MDS/AML emerged as a common finding in patients with AA. Prior studies reported that mutations in PIGA and BCOR had a better response to immunosuppressive therapy and improved clinical outcome, specifically improved progression‐free survival (PFS) and overall survival (OS), while AA patients with mutated ASXL1, DNMT3A, JAK2/JAK3, or RUNX1 fare worse with regard to immunosuppressive therapy response and survival [7, 8]. A high-risk clonal evolution was detected in AA patients with ASXL1, DNMT3A or RUNX1 mutations [8,9,10]. However, recent studies demonstrated that mutations did not correlate with hematologic response and OS, either at baseline or in new or additional mutations; treating physicians should not overinterpret the presence of mutant clones at diagnosis or after therapy in isolation, and detection of these clones do not warrant high-risk procedures, such as hematopoietic stem-cell transplantation [2, 11].
Thus far, the impact of somatic mutations in AA on clinical and hematological outcomes is not completely established. Therefore, we conducted this study using bone marrow samples of AA patients in targeted sequencing to investigate the frequencies of somatic mutations in AA and their correlation with prognostic relevance and response to immunosuppressive treatment.
Materials and methods
Patients
We investigated a cohort of 279 hospitalized AA patients from the hematology department of the First Affiliated Hospital of Zhengzhou University between January 2016 and June 2021. At the time of enrollment, we excluded patients without completion of next-generation sequencing. The following laboratory and clinical information were obtained for each patient: blood cell count, ferritin, iron and the somatic mutations of bone marrow cells, age, sex, date of diagnosis and treatment. Furthermore, patients were followed up till September 2022 for disease progression, survival and response to treatment. AA patients were diagnosed according to 2016 British criteria, and the inherited AAs were excluded. [12] The severity of AA was graded according to the blood count parameters and bone marrow findings. Severe AA (SAA) was defined as BM cellularity < 25%, or 25–50% with < 30% residual hematopoietic cells and at least two of the following: (I) absolute neutrophil count < 0.5 × 109/L, (II) platelets < 20 × 109/L and (III) reticulocyte count < 20 × 109/L. AA patients who did not meet the criteria for SAA were classified as non-severe AA (NSAA). The treatment outcome was based on previous literature [4, 11, 13]. Overall survival (OS) was defined as the time from enrollment to death from any cause or the last follow-up. Event-free survival (EFS) was defined as the first event time from diagnosis to the last follow-up. Event: clonal evolution (MDS/AML), relapse, death, and progression to severe AA. For comparison, we included a cohort of 174 MDS patients who performed targeted sequencing during the same period, diagnosed according to the 2016 World Health Organization classification [14]. To better compare with AA patients, we screened MDS from patients with pancytopenia.
The inclusion criteria were: (1) age ≥ 14 years old, (2) NGS was performed and (3) complete clinical data.
Gene sequencing
Gene mutation analysis was performed by NGS of the DNA samples extracted from the bone marrow monocytes in patients. The NGS libraries were paired-end sequenced (2 × 150 bp) on an Illumina MiSeq System (Illumina, San Diego, CA). The mean depth of each sample was 2500 × , with an average of 98% of the target sequence covered sufficiently deep for variant calling. Detection sensitivity was ~ 5% (a mutation with 5% or more variability can be reported).
The Illumina MiSeq System (Illumina, San Diego, CA) is a high-throughput sequencing platform based on Sequencing by Synthesis (SBS) technology and sequence libraries to produce large amounts of high-quality data. Analyses were conducted of the relevant mutations of 22 genes, ASXL1, NPM1, KIT, FLT3, CEBPA, DNMT3A, IDH1/2, TET2, EZH2, RUNX1, PHF6, TP53, SF3B1, SRSF2, U2AF1, ZRSR2, NRAS, CBL, SETBP1, ETV6, and JAK2. Each mutation was analyzed in the Catalog of Somatic Mutations in Cancer databases (COMSIC; https://cancer.sanger.ac.uk/cosmic/). All bone marrow samples were collected with informed consent, and the study was reviewed and approved by the First Affiliated Hospital of Zhengzhou University College of Medicine.
Statistical analysis
Continuous variables were described by median and range, while classification variables were described by example and percentage. Mann–Whitney U test was used for continuous variables, and the chi-square test and Fisher exact test were used for classified variables. The OS and EFS were estimated using the Kaplan–Meier curve and compared by the Log-rank test. The logistic regression model was used for single and multivariate analyses, and the variables with P < 0.1 were included in the multivariate analysis. The statistical analyses were performed using SPSS version 20.0. GraphPad Prism 8 was run to generate graphs. A P-value of < 0.05 was considered statistically significant.
Results
Somatic mutation
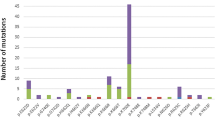
A total of 279 AA patients were enrolled in this study, including 128 females (45.9%) and 151 males (54.1%), with a median age of 39(14–85) years at diagnosis. Targeted NGS of a panel of genes that were recurrently mutated in myeloid cancers was performed using bone marrow obtained from the patients. Overall, there were 25 patients (9.0%) with somatic mutations, 20 patients (7.2%) had one mutation, four patients (1.4%) had two mutations, and one patient (0.4%) had more than two mutations (Fig. 1 Frequency of Mutation in the AA and MDS groups). The most frequently mutated genes were ASXL1(in 2.9% of the patients), DNMT3A (1.8%) and TET2 (1.8%) (Fig. 2 Somatic Mutations Identified by next-generation sequencing and Supplementary Table 1). Details of the somatic mutations in AA patients are given in Supplementary Table 2. Moreover, there were 174 MDS patients, 112 males (64.4%), and the median age at diagnosis was 55 (15–87) years. Analysis of mutation results, at least one gene mutation was detected in 120 patients (69.0%), including 39 patients (22.4%) who had one mutation, 48 patients (27.6%) had two mutations, and 33 patients (18.9%) had more than two mutations (Fig. 1). The most frequently mutated genes were U2AF1 (in 24.7% of the patients), ASXL1 (18.4%) and TP53 (13.2%) (Fig. 2 and Supplementary Table 1). The number of mutations per patient was lower in AA than in MDS, typically one mutation per patient compared with a median number of two in MDS. There was a statistically significant difference in the number of mutations between the two groups (Fig. 1). At the same time, the gene type and frequency of each gene mutation type were fewer than in the MDS group (Fig. 2 and Supplementary Table 1). Furthermore, the median variant allele frequency (VAF) in AA was substantially lower than that of patients with MDS (6.9% vs. 28.4%) with statistical significance (Fig. 3 Comparison of the median variant allele frequency between AA and MDS cohorts). Similarly, the median VAF of each somatic mutation was lower in AA patients than MDS, as shown in Fig. 4 (Comparison of variant allele frequencies individual mutation between the AA and MDS cohorts). A comparison of the mutation of patients in various age groups showed that the proportion of mutation in MDS patients increased with age. The incidence of gene mutation in the AA group increased with the trend, but there was no statistical significance (Supplementary Fig. 1 Prevalence of somatic mutations, according to age).

The bar chart shows the frequency and number of mutations in AA and MDS. Blue color indicates the frequency of patients with one mutation (7.2% in AA or 22.4% in MDS), blue indicates the frequency of patients with two or more mutation (1.8% in AA or 46.6% in MDS)

Bar chart showing the frequency of mutated genes and type of mutations in each gene identified in the AA and MDS groups

Box figure show the median variant allele frequency in the AA and MDS cohorts. The box-and-whisker plots of the specific gene mutations are shown; the whiskers indicate the range, the sides of the boxes indicate the interquartile range, and the vertical line within each box indicates the median(6.9% vs. 28.4%). P values were obtained using Wilcoxon rank sum test

Box figure show the variant allele frequency of individual mutations in the AA and MDS cohorts. The box-and-whisker plots of the specific gene mutations are shown; the whiskers indicate the range, the sides of the boxes indicate the interquartile range, and the horizontal line within each box indicates the median
Clinical correlations
Patients were classified into two groups by mutation: 25 patients with a somatic mutation (SM, 9.0%) and 254 patients with no somatic mutation (N-SM, 91.0%). In the SM group, the median age was 53 (14–85) years, significantly higher than the N-SM group, with a median age of 38 (14–77) years (P < 0.05, Table 1). Among 279 patients with AA, we excluded five patients who had transplant within 3 months, and 29 patients were lost to follow-up. Finally, we enrolled 245 patients to assess treatment response, prognosis and the relationship between somatic mutation, consisting of the SM group (n = 24, 9.8%) and the N-SM group (n = 221, 90.2%). The median age of the patients in the SM group was higher than in the N-SM group (P = 0.012). Comparing laboratory examination, we found that the SM group had a higher monocyte count than the N-SM group, with the values of 0.18 (0.02–0.89) × 109/L vs. 0.13 (0.00–0.75) × 109/L. There was a statistically significant difference between the two groups (P < 0.05, Table 2). However, other blood parameters were not significantly different between SM patients and N-SM patients. There were no significant differences between patients with or without mutations in the severity of AA (P = 0.785), cytogenetics (P = 0.164) and treatment (P = 0.564).
AA patients were subdivided into three groups according to treatment: anti-thymocyte globulin combined with or without cyclosporine/thrombopoietin-receptor agonist (Eltrombopag /Herombopag), cyclosporine combined with or without thrombopoietin-receptor agonist (Eltrombopag /Herombopag), and others (supportive or other immunosuppressive therapy). Furthermore, we included the efficacy of treated AA patients for three and 6 months to assess the potential impact of somatic mutations on the treatment response. In the SM group, the overall response to IST at three and 6 months were 37.5% and 66.7%, respectively, and there was no significant difference compared with the N-SM group (P > 0.05, Table 1). Additionally, we included the factors that might affect treatment in logistic, univariate and multivariate analyses. The results showed that mutation, reticulocyte, absolute neutrophil count and lymphocyte count did not affect the efficacy(P > 0.05,); only the platelet count and the treatment options affected the treatment response at 3 months (P < 0.05, Tables 3, 4). Surprisingly, ferritin influenced the 3-month overall response of patients in univariate analysis. Similar results were found for the influence factor of overall response at 6 months (P < 0.05, Table 3).
Longer-term follow-up
Among the 245 AA patients, the median follow-up was 25 months (95% confidence interval [CI] 25.3–28.8) in all patients. The median follow-up among the patients in SM and N-SM groups was 22 months (95% CI 19.2–32.1) and 26 months (95% CI 25.4–29.0). During the entire follow-up period, four (16.7%) patients in the SM group and 44 (19.9%) patients in the N-SM group died. By comparing the survival curves of the mutant and non-mutant groups, we found that the presence of somatic mutations had no significant effect on the AA patients OS (Fig. 5 Overall survival correlations with somatic mutations). In the SM group, five (20.8%) patients had an event, while 48 (21.7%) patients had an event in the N-SM group, Kaplan–Meier curves for EFS were not significantly different (Fig. 6 Event-free survival correlations with somatic mutations). At the same time, we also compared the OS and EFS of ASXL1 and DNMT3A mutant and non-mutated groups, which was not statistically significant (Fig. 7 Overall survival correlations with ASXL1/DNMT3A somatic mutation and Fig. 8 Event-free survival correlations with ASXL1/DNMT3A somatic mutation).

Kaplan–Meier curves for overall survival among patients with and without somatic mutation. Log-rank tests were used for statistical comparisons among the groups(p = 0.54)

Kaplan–Meier curves for event-free survival among patients with and without somatic mutation. Log-rank tests were used for statistical comparisons among the groups (p = 0.82)

Kaplan–Meier curves for overall survival among patients with and without ASXL1/DNMT3A somatic mutation. Log-rank tests were used for statistical comparisons among the groups (p = 0.45)

Kaplan–Meier curves for event-free survival among patients with and without ASXL1/DNMT3A somatic mutation. Log-rank tests were used for statistical comparisons among the groups (p = 0.07)
Three patients who were older than 50 years had clonal progression to MDS at four, 4, and 9 months, respectively, after immunosuppressive therapy. Moreover, there were three cases of secondary MDS patients without somatic mutation before transformation and the normal chromosome karyotype. Two had no response at 6 months, while the third had a partial response. After transformation, one patient had a karyotype change of 46XX, − 7 (− 7/7q−), and one patient had ASXL1 mutation, both of them died at 9 and 17 months, respectively. One patient carrying ASXL1, SETBP1 and RUNX1 mutations evolved to AML within seven years of the AA diagnosis; this patient was diagnosed with NSAA at the age of 47 and received oral cyclosporine therapy for two years, but the disease relapsed after discontinuation of the drug. Moreover, there was no durable response upon subsequent cycles of immunosuppressive therapy. The cytogenetic karyotype at the time of evolution was 46XY, − 7, + 21[7]/46XY [3].
Discussion
With the advancement of NGS, somatic mutations associated with myeloid malignancies have been found in AA patients. Somatic mutations are associated with clonal hematopoiesis in AA, affecting the treatment and survival and further clonal evolution into MDS/AML. In the present study, 279 AA patients were included, and MDS was used as a control to clarify the proportion of somatic mutations in AA patients in our hospital. In addition, we followed up with 245 patients with AA, analyzing the influence of the somatic mutation on treatment and survival to help doctors make treatment decisions.
In our study, 9% of AA patients had myeloid tumor-related somatic mutations. Similarly, Daria V Babushok et al. reported mutations only in a small subset of patients with AA (9%), and Heuser M et al. identified three somatic mutations in two patients in the examined MDS candidate genes (5.3%). [15, 16] However, compared with most studies that reported somatic mutations in about 21% of patients, mutation frequency in our study was significantly lower, which could be due to the small number of gene panel for targeted sequencing and the absence of the most common mutated genes in AA: PIGA and BCOR. [7, 8, 10, 17, 18] The most commonly mutated genes in AA were ASXL1, DNMT3A and TET2. After excluding PIGA and BCOR mutations, the most frequently mutated genes were ASXL1, DNMT3A and TET2 in AA patients, similar to the results of our study. [10, 19, 20] The mutation pattern in AA patients were different from the MDS patients. In our study, the most frequently mutated genes in MDS patients were U2AF1, ASXL1 and TP53. It is worth noting that SF3B1 mutation frequency was pretty low compared with previous studies [21]. The presumed reasons are as follows. First, SF3B1 mutations were mainly found in MDS patients with ring sideroblasts, and the hemogram of these patients generally presented decreased hemoglobin [22]. We excluded patients with anemia alone, which may lead to a lower SF3B1 mutation in the MDS cohort. Second, it may be due to the small sample size of our study. Although ASXL1 and DNMT3A mutations were frequent in AA, MDS and AML, there is a substantial underrepresentation of mutations in splicing-factor genes, JAK2, and TP53 in AA compared with MDS and AML, reflecting the difference in the mechanism of clonal selection between both diseases and mutations. [3, 23,24,25] Meanwhile, our data showed that the median allelic burden of mutations in AA was substantially lower than that in MDS (6.9% vs. 28.4%). The low-burden mutations might be transient events and might not contribute to later evolution. However, along with somatic chromosomal aberrations, the mutational burden (total volume of acquired mutations) might serve as a measure of genomic damage. [26]
Benign clonal hematopoiesis identified in healthy individuals is known as clonal hematopoiesis of indeterminate potential(CHIP). The somatic mutations that drive CHIP were most frequently involved in DNMT3A and TET2, which are also more frequent in AA. The difference in somatic mutations between AA patients and CHIP lesion populations remains to be explored. DNMT3A and TET2 mutations in AA patients might represent CHIP lesions. In AA, somatic mutations become detectable as the contraction of the stem cell compartment leads to decreased hematopoietic stem cells. However, concerning previous literature, we have different speculation. Research showed that the frequency of mutations increases in frequency with age in the general population. Mutations in genes implicated in hematologic cancers were found in < 1% of healthy persons younger than 40 years of age, 1.7% of persons 40–49 years of age, 2.5% of persons 50–59 years of age, 45.2% of persons 60 years of age or older [27]. The mutations in DNMT3A and TET2 comprised 57.2% and 33.3% of all recorded mutations in CHIP lesions, respectively [28]. Furthermore, the prevalence of TET2 mutations in individuals 55 years of age or older showed a consistent increase with age, averaging 6.8% per year [29]. In our study, somatic mutations were observed in 5.9% of patients younger than age 40 years, 12.2% in those 40–49 years, 16.7% in those 50–59 years, and 24.8% of patients 60 years of age or older. DNMT3A and TET2 mutations accounted for the same proportion of all documented mutations in AA patients, which was 16.1% (5/31). Moreover, when the patient was younger than 60 years old, the frequency of somatic mutations in AA patients in our study was higher than that in previous studies of CHIP lesions. Additionally, the mutation frequencies of DNMT3A and TET2 were different in AA and CHIP patients. Finally, although somatic mutations did not affect the treatment response and survival of AA patients in our study, Huang et al. [8] reported that AA patients with TET2 mutations had a better response to IST than unmutated ones, and Park et al. [18] suggested DNMT3A mutations was an adverse factor associated with short overall survival. In conclusion, we speculate that the DNMT3A and TET2 mutations in AA patients may be different from those in CHIP lesions, but further follow-up is needed for verification.
The somatic mutations were a common finding in the elderly [30]. According to our data, SM patients were older than N-SM patients, but the mutation frequency does not increase markedly with age. On the one hand, it is possible that with aging, there is a stereotypical outgrowth of cells bearing mutations in epigenetic regulators of stem cell renewal, DNMT3A, TET2, and ASXL1, as well as others [31]. This age-dependent background of stochastic mutations also shapes clonal diversity in AA, as hematopoietic and progenitor cells with various background mutations “struggle for existence” and may derive a selective advantage from their somatic differences [17]. Indeed, AA cases presenting at an older age may already carry somatic age-related mutations, which could be positively selected after multiple rounds of IST because of the inherent increased fitness advantage of oligoclonal over normal hematopoietic stem and progenitor cells.[20] On the other hand, it is considered that somatic mutations may occur during the pathogenesis of AA. The depletion of AA stem cell pool was conducive to the growth of some hematopoietic stem cells with somatic mutations. These hematopoietic stem cells through acquisition of somatic alterations, becomes either less immunogenic or acquires relative resistance to cytotoxicity or cytokine-mediated marrow suppression, were more likely to survive [32, 33]. Babushok et al. showed that clonal hematopoiesis can occur early in the course of the disease even in young patients with AA, suggesting that somatic mutations in AA are not always associated with aging [15].
As for the relationship between somatic mutations and blood cell counts, studies have reported that red-cell distribution width is significantly increased in patients with somatic mutations and associated with all-cause death in patients. [27] At the same time, some studies indicated that AA patients with somatic mutations had higher neutrophil counts. [11] We analyzed the blood routine of the two groups and found that the monocyte count of the mutation patients was significantly higher, but there was no significant difference in red-cell distribution width and absolute neutrophil count between the two groups.
A long-term follow-up of 245 AA patients was conducted to observe the efficacy of IST treatment at 3 and 6 months. The results showed that somatic mutations did not affect the treatment response of AA patients. This is consistent with another research stating no significant differences in OS and response to IST between patients with somatic mutations and without mutations [10, 11, 34]. Previous studies reported that patients with ASXL1/DNMT3A mutations had poor immunosuppressive responses and shorter survival [7, 18]. In addition, we analyzed the response and survival of patients with ASXL1 and DNMT3A mutations and without mutations, and the results showed no significant differences in both groups. Furthermore, new or additional mutations after treatment were not predictive of a lack of response or other dire outcomes. [11] Univariate and multivariate logistic regression displayed that the treatment regimen affected the response at three and 6 months. The somatic mutations and previously reported hematologic characteristics at baseline (absolute reticulocyte count and absolute lymphocyte count) were not associated with the overall response rate in our trial [35]. Surprisingly, platelet count affected the 3-month efficacy of AA patients, and ferritin influenced the 3-month overall response of patients in univariate analysis and the 6-month overall response.
Retrospective studies with long follow-up revealed that 10–20% of patients with AA treated with IST developed either MDS or AML, and patients of older age at diagnosis, a shorter telomere length, poor response to standard IST and longer disease duration were at higher risk of clonal evolution [5, 17, 20, 30]. Recently, in a cohort of 407 patients from the National Institutes of Health, the presence of specific somatic mutations was found to be predictive of evolution to MDS/AML when detected at 6 months after IST; including RUNX1, splicing factor mutations, and ASXL1.[9] By contrast, the predictive value of isolated mutations in genes like TET2 and DNMT3A, which are frequently mutated in age-related clonal hematopoiesis was lower, these genes require additional genetic events to give rise to a myeloid neoplasm. [27, 36, 37] In our study, there were four cases of clonal progression; all were older than 50 years, three cases progressed to MDS with normal chromosomal karyotype and no detectable somatic mutations at the time of the sequence analysis before diagnosis, and one case progressed to AML with three somatic mutations, ASXL1, SETBP1 and RUNX1, respectively. Overall, because of the rarity of AA and the long latency of clone evolution, the contribution of mutations to the risk of malignancy in AA patients is less clear, necessitating longer follow-up and larger patient numbers, and should be considered within the context of other patient and disease-specific factors, as well as the function of the mutated gene in hematopoiesis, aging, and autoimmunity [6, 34].
In conclusion, our data indicated that myeloid tumor-associated somatic mutations in AA patients were detected in only a minority of patients by NGS sequencing. AA and MDS patients had different gene mutation patterns. The somatic mutations in patients with AA were characterized by lower mutation frequency, mostly one mutation, and lower median allelic burden of mutations than MDS. Somatic mutations were a common finding in the elderly, and the frequency of mutations increases with age. The platelet count affected the response to treatment at 3 months, and ferritin level affected the outcome at 6 months, while somatic mutations were not associated with treatment response or long-term survival. Nevertheless, since our cohort of patients with the mutation was small, this result needs to be further confirmed with larger series of patients.
References
Young NS. Aplastic anemia. N Engl J Med. 2018;379:1643–56.
Scheinberg P. A new standard immunosuppression regimen in severe aplastic anemia. N Engl J Med. 2022;386:89–90.
Durrani J, Groarke EM. Clonality in immune aplastic anemia: Mechanisms of immune escape or malignant transformation. Semin Hematol. 2022;59:137–42.
Bacigalupo A. How I treat acquired aplastic anemia. Blood J Am Soc Hematol. 2017;129(11):1428–36.
Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130–5.
Sun L, Babushok DV. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood. 2020;136:36–49.
Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47.
Huang J, Ge M, Lu S, et al. Mutations of ASXL1 and TET2 in aplastic anemia. Haematologica. 2015;100:e172–5.
Groarke EM, Patel BA, Shalhoub R, et al. Predictors of clonal evolution and myeloid neoplasia following immunosuppressive therapy in severe aplastic anemia. Leukemia. 2022;36:2328–37.
Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–704.
de Peffault LR, Kulasekararaj A, Iacobelli S, et al. Eltrombopag added to immunosuppression in severe aplastic anemia. N Engl J Med. 2022;386:11–23.
Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187–207.
Sasaki N, Shimura K, Yoshida M, et al. Immunosuppressive therapy with rabbit antithymocyte globulin therapy for acquired aplastic anemia: a multi-institutional retrospective study in Japanese adult patients. Int J Hematol. 2019;109:278–85.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Babushok DV, Perdigones N, Perin JC, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015;208:115–28.
Heuser M, Schlarmann C, Dobbernack V, et al. Genetic characterization of acquired aplastic anemia by targeted sequencing. Haematologica. 2014;99:e165–7.
Babushok DV. A brief, but comprehensive, guide to clonal evolution in aplastic anemia. Hematology. 2018;2018:457–66.
Park HS, Park SN, Im K, et al. Telomere length and somatic mutations in correlation with response to immunosuppressive treatment in aplastic anaemia. Br J Haematol. 2017;178:603–15.
Shallis RM, Ahmad R, Zeidan AM. Aplastic anemia: etiology, molecular pathogenesis, and emerging concepts. Eur J Haematol. 2018;101:711–20.
Gurnari C, Pagliuca S, Prata PH, et al. Clinical and molecular determinants of clonal evolution in aplastic anemia and paroxysmal nocturnal hemoglobinuria. JCO. 2022. https://doi.org/10.1200/jco.22.00710.
Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): An update. Mutat Res Rev Mutat Res. 2016;769:47–62.
Tang Y, Miao M, Han S, et al. Prognostic value and clinical feature of SF3B1 mutations in myelodysplastic syndromes: a meta-analysis. Crit Rev Oncol Hematol. 2019;133:74–83.
Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337–47.
Negoro E, Nagata Y, Clemente MJ, et al. Origins of myelodysplastic syndromes after aplastic anemia. Blood. 2017;130:1953–7.
Boddu PC, Kadia TM. Molecular pathogenesis of acquired aplastic anemia. Eur J Haematol. 2019;102:103–10.
Maciejewski JP, Balasubramanian SK. Clinical implications of somatic mutations in aplastic anemia and myelodysplastic syndrome in genomic age. Hematology. 2017;2017:66–72.
Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130:753–62.
Fabre MA, de Almeida JG, Fiorillo E, et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature. 2022;606:335–42.
Mufti GJ, Marsh JCW. Somatic mutations in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:595–607.
Gibson CJ, Steensma DP. New insights from studies of clonal hematopoiesis. Clin Cancer Res. 2018;24:4633–42.
Stanley N, Olson TS, Babushok DV. Recent advances in understanding clonal haematopoiesis in aplastic anaemia. Br J Haematol. 2017;177:509–25.
Cooper JN, Young NS. Clonality in context: hematopoietic clones in their marrow environment. Blood. 2017;130:2363–72.
Winkler T, Fan X, Cooper J, et al. Treatment optimization and genomic outcomes in refractory severe aplastic anemia treated with eltrombopag. Blood. 2019;133:2575–85.
Scheinberg P, Wu CO, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol. 2009;144:206–16.
Malcovati L, Gallì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129:3371–8.
Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood dna sequence. N Engl J Med. 2014;371:2477–87.
Acknowledgements
The authors acknowledge the team of researchers at the Clinical Laboratory Center Institute of Department of Hematology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China, for their professional assistance. The author is grateful to the patients for participating in research studies of somatic mutations in aplastic anemia. This work was supported by a grant from Science and Technology Department of Henan Province (SBGJ202102150, recipient Y.L.; LHGJ20200332, recipient D.Z.)
Funding
This work was supported by a grant from Science and Technology Department of Henan Province (SBGJ202102150, recipient Y.L.; LHGJ20200332, recipient D.Z.)
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. YL designed and directed the study. LL and DZ wrote the manuscript draft. LL, DZ, QF, JW, JY, DC collected data. FW, RG, XX and ZJ provided resources. All authors critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval and Consent to participate
This study has been approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University, in accordance with the Declaration of Helsinki.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, L., Zhang, D., Fu, Q. et al. Clinical implications of myeloid malignancy‑related somatic mutations in aplastic anemia. Clin Exp Med 23, 4473–4482 (2023). https://doi.org/10.1007/s10238-023-01067-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-023-01067-4