
Abstract
Objectives
Medical devices are potentially good candidates for coverage with evidence development (CED) schemes, as clinical data at market entry are often sparse and (cost-)effectiveness depends on real-world use. The objective of this research was to explore the diffusion of CED schemes for devices in Europe, and the factors that favour or hamper their utilization.
Methods
We conducted structured interviews with 25 decision-makers from 22 European countries to explore the characteristics of existing CED programmes for devices, and how decision makers perceived 13 pre-identified challenges associated with initiating and operating CED schemes for devices. We also collected data on individual schemes that were either initiated or still ongoing in the last 5 years.
Results
We identified seven countries with CED programmes for devices and 78 ongoing schemes. The characteristics of CED programmes varied across countries, including eligibility criteria, roles and responsibilities of stakeholders, funding arrangements, and type of decisions being contemplated at the outset of each scheme. We observed a high variability in how decision makers perceived CED-related challenges possibly reflecting country-specific arrangements and different experiences with CED. One general finding across all countries was that relatively little attention was paid to the evaluation of schemes, both during and at their completion.
Conclusions
CED programmes for devices with different characteristics exist in Europe. Decision-makers’ perceptions differ on the challenges associated with these schemes. More exchange of knowledge and experience will help decision makers anticipate the likely challenges in CED schemes for devices, and to learn from good practices existing elsewhere.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
At the time of the publication of the ISPOR ‘Good Practices for Performance-Based Risk-Sharing Arrangements (PBRSAs) Task Force’ report [1], it was acknowledged that there were two types of arrangements to aid the market entry of new technologies; finance-based and performance-based agreements. Briefly, in finance-based arrangements, agreements between payers and manufacturers are purely financial and may involve for example price–volume agreements, price discounts or budget caps. In PBRSAs one of the key elements is that the price, or reimbursement of a technology is linked to its performance which is assessed through a purposeful, prospective data collection.. Indeed, some of the earliest examples of PBRSAs concern coverage with evidence development (CED) schemes that were initiated by the Centres for Medicare & Medicaid Services in the US and the Ontario Ministry of Health in Canada for medical procedures and devices [2, 3]. In CED schemes, data are collected with the objective of reducing uncertainty concerning the clinical or cost-effectiveness of a health technology and to assist in future decisions about its reimbursement, coverage, or recommendations for its use. Typically, these schemes are centrally coordinated and require substantial data collection.
Since the publication of the Task Force’s report, two trends can be observed. Firstly, there has been a growth in the popularity of finance-based agreements, or simple price reductions, as compared with performance-based schemes. In a recent review of managed entry agreements in Europe, Dabbous et al. [4] note that ‘despite the interests in CED schemes, European countries have moved towards finance-based agreements due to the complexities and burdens associated with PBRSAs’. The lack of appetite for complex agreements among policy-makers was also noted by Karlsberg Schaffer et al. [5], who concluded that ‘there is a mismatch between the enthusiasm in the academic literature for developing new approaches and the scepticism of payers that they can work, or are necessary for the foreseeable future’. Secondly, there has been a growth in the application of financed-based and performance-based agreements to drugs rather than to other types of technologies, which could be a response to the growing number of transformational, but highly expensive new drugs entering the market [6]. Many recent reviews of PBRSAs discuss issues that apply to health technologies in general but draw almost exclusively on drugs for their examples [4, 7,8,9].
In principle, medical devices are good candidates for PBRSAs, particularly for CED schemes, since there are often considerable uncertainties concerning their (cost-)effectiveness. This is mainly because the data requirements to obtain market access are often less stringent than those for drugs, and therefore devices are generally adopted in clinical practice with relatively little clinical or economic evidence [10, 11].. In contrast to pharmaceuticals where the market authorization and supervision is centrally managed by the European Medicine Agency (EMA) (Regulation (EC) No 726/2004), the conformity assessment procedures for medical devices of risk class II or higher in Europe are decentralized and operated by public or private notified bodies (NBs) which are designated by the EU member states. Evidence requirements for market authorization are regulated by the medical device regulation (MDR), which also defines when a clinical investigation of the new device is required or when conformity assessment can be based on the equivalence principle with a previously marketed device. However, notwithstanding the requirements for clinical investigations, a controlled clinical trial, which demonstrates the relative effectiveness compared to alternative treatments, is generally not mandatory for MDs. Besides the differences in the regulatory approaches compared with pharmaceuticals, certain sources of uncertainty around a medical device are relatively less easy to explore by means of pre-market studies. Many devices are part of complex interventions, consisting of multiple behavioural, technological, and organizational components, and therefore their actual (cost-)effectiveness profile usually depends on a series of context-specific factors that are difficult to assess before their adoption in the real-world. For example, device performance in regular clinical practice often depends not only on the device itself, but also on the skills of the user [12, 13]. In addition, while finance-based agreements are also possible, the cost of adopting a new device depends not only on its price, but also the cost of any new procedures or other organizational changes that might be required for its use. Therefore, a price reduction for the device itself may have less of an impact on overall costs. Moreover, finance-based agreements do not resolve potential issues about uncertainty in the effectiveness of the device, which both payers and patients may feel is important.
The pace of innovation in medical devices is considerable, with many new products entering the market every year. For example, in 2017, the number of patents in the field of medical technologies filed with the European Patent Office (EPO) was more than double compared to the number concerning pharmaceuticals (13,000 versus 6300), and the total expenditure on medical technologies in Europe was roughly estimated as €115 billion [14]. Given the relevance of the market and the above-mentioned challenges with evidence generation at market launch, any policy tool such as CED, which foresees a controlled introduction of a technology while collecting further post-market evidence, is highly relevant in the context of medical devices. However, despite the possible advantages of CED schemes for aiding coverage decisions regarding new devices, little is known about the extent to which these schemes are used in Europe and the detailed perceptions of decision-makers regarding their utilization [15,16,17]. Therefore, the objective of this research was to contribute to filling this gap by exploring the characteristics and diffusion of CED schemes for devices in Europe, and the challenges that decision-makers face during the different phases of a scheme [1, 14]. Our aim was to assist those considering the implementation of CED schemes for medical devices and to increase the understanding of both how schemes are currently being applied in Europe and how the challenges associated with them are being addressed.
Methods
This study is part of the EU Horizon 2020 COMED project that has been reviewed and approved by the Bocconi University Ethics Committee (protocol number: 0068538, approved on May 8, 2018).
The research was conducted in three consecutive steps: (1) development of a structured interview guide (2) interviews with decision-makers from a sample of European countries, (3) synthesis and qualitative content analysis of the interview data, the data made available by the decision-makers during or following the interview, and data on scheme characteristics previously obtained [17]. The steps are described in more detail below.
Development of the interview guide
We developed a structured interview guide (Online Resource 1) that consisted of three sections. Section A included general questions on whether CED programmes underpinning the individual schemes existed in the decision-maker’s country and for which type of technology they were used. Section B included questions on 13 challenges for CED schemes for devices (Table 1). This list was derived from a recent systematic review that identified 20 challenges for CED schemes for devices [17]. To reduce the participants’ burden, we reduced the original list of 20 challenges to 13, by grouping different aspects of the same general challenge. The final list of challenges was discussed and agreed among all authors to ensure that all relevant aspects originally identified were covered in the interview guide (see the Online resource 2 for more details).
We asked the decision-makers to assess how they perceived the 13 challenges to apply to CED schemes for devices on a six-point Likert scale (ranging from 0 “not a challenge” to 5 “a major challenge”). Where CED schemes for devices existed, we also asked respondents how the challenges were met in their country, and the interview proceeded to Section C. Otherwise, the interview ended here. Section C included questions on the detailed characteristics of individual CED schemes for devices that had been either initiated or still ongoing in the past five years. These questions concerned a description of the device under evaluation, its clinical application, the objective of the scheme, key sources of uncertainty, funding of the scheme, its design, the decision rule, and outcome (if re-assessment was done), and any public source of information on the scheme.
Interviews with decision-makers
A first draft of the interview guide was circulated for comments among the COMED project partners. Subsequently the final draft of the interview guide was pilot tested during interviews with one Italian policy maker and two academic experts with extensive experience of CED in Canada and the USA, two countries with a substantial number of schemes.
The interviews were conducted face-to-face or by telephone between June and December 2019. Decision-makers from decision bodies at the central (or in two cases regional) level were identified from the professional networks of the members of the COMED project team or the websites of relevant decision bodies in the following European countries: Austria, Belgium, Bulgaria, Croatia, Czech Republic, Denmark, England, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, the Netherlands, Norway, Poland, Portugal, Romania, Scotland, Slovakia, Slovenia, Spain, Sweden, and Switzerland. Other countries from the EU/EEA were excluded because it was not possible to identify a relevant decision-making body for the technology assessment of medical devices. We invited decision-makers to participate in the study by sending them an email with information on the COMED project and the objective of our study. When we were unable to identify a decision-maker from the networks or websites, we sent the information and invitation to the relevant decision bodies. In three cases where no relevant decision-maker could be identified (i.e. Bulgaria, Czech Republic, and Sweden), we invited academic researchers with relevant expertise to participate. None of these countries had however any CED programme for devices in place. We interviewed more than one decision maker from a given country in cases where schemes were operated in more than one jurisdiction (i.e., Italy), where more than one decision body was involved in operating schemes (i.e., France), or where more than one decision-maker, from different parts of the relevant organization, agreed to participate (i.e., England). We excluded Croatia, Iceland, Romania, and Slovenia from our sample after repeated attempts to schedule an interview by December 2019 were unsuccessful. Information on the individual CED schemes provided by decision-makers during or following the interview was supplemented with information on individual schemes previously obtained [17], compiled in tabular form, and sent to the participants for a validity check.
Data analysis
The transcripts were subjected to qualitative content analysis using deductive coding to meet the objective of this research. The results of each interview were reported in a table by one author (CF) and assessed by two authors (CF and VRD) who independently extracted the relevant information. Agreement on the data to be reported was then reached through discussion and further analysis of the original transcripts. The data obtained from Sections A and C of the interview guide, together with the data obtained prior to and following the interviews were used to identify and classify the characteristics of the existing CED programmes for devices according to the four phases of CED schemes: 1) assessing the desirability of the scheme; 2) designing the scheme; 3) implementing the scheme, and 4) evaluating it (1). These phases are described in more detail in Table 2. The information collected was then synthesised in a narrative review.
The data obtained from Section B of the interview guide were used to obtain insight into the participants’ perceptions of the 13 challenges and into the factors that influenced their score for a particular challenge. The quantitative data obtained from Section B were used to calculate the mean (SD) and median (IQR) Likert scores for the 13 challenges (excluding the challenges that were marked as ‘not applicable’ by the participants). Then, we calculated these statistics separately for participants from countries with and from countries without a CED programme for medical devices. Because of the small sample sizes, we did not examine the differences in scores by performing statistical tests, but all factors which were perceived as having a positive or negative influence on each challenge were synthetized in tabular form.
Results
We interviewed 25 participants from 23 jurisdictions. Respondents were from national or regional health authorities (n = 15); national health insurance bodies (n = 2); hospitals (n = 3); and universities (n = 3) (see Online Resource 3 for details). Eighteen participants had high-level managerial roles related to the HTA of medical devices or services, or were responsible for the CED programme in their jurisdiction; four participants were technical advisers directly involved in the assessment of medical devices, and three were academics with an expertise in conditional reimbursement schemes. In seven out of the 23 jurisdictions (30.4%), CED programmes existed that included (or were specific to) schemes for medical devices (i.e., Belgium, England, France, Germany, the Netherlands, Spain, and Switzerland). In France, two different programmes were identified: Post Registration Studies (PRS) for devices submitting for registration into the positive list of reimbursable products and services (LPPR list); and Forfait innovation (FI) for highly innovative technologies early in their development phase. Of the remaining jurisdictions, 5 (21.7%) operated CED programmes for drugs only (i.e., Bulgaria, Hungary, Portugal, Scotland, and Slovakia), and 11 (47.8%) did not operate any CED programmes (i.e., Austria, Czech Republic, Denmark, Finland, Greece, Ireland, Italy-Emilia Romagna Region, Italy-national level, Norway, Poland and Sweden), although some of these may have other types of PBRSAs such as performance linked reimbursement schemes (e.g., payment by results schemes). In addition, single ‘one-off’ experiences with schemes for specific devices were reported by participants from Emilia Romagna Region in Italy and Ireland, in the absence of formal programmes for CED schemes for devices.
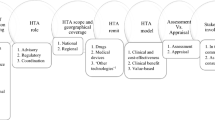
Overall, we identified 78 CED schemes for devices which were ongoing in the last 5 years in Europe. A full overview of the characteristics of these schemes is included in Online Resource 4. Table 3 and Fig. 1 present an overview of how the existing national CED programmes underpinning the individual schemes address the different phases of CED schemes. Our main findings are highlighted below.

Overview of the main characteristics of CED programmes in Europe
Assessing the desirability of a CED scheme
We identified three main ways in which devices are selected for a scheme (Table 3). Firstly, a device can be selected as the direct result of a formal health technology assessment (HTA), if the decision body making the assessment identifies remaining uncertainties on the device (cost-)effectiveness and therefore propose initiation of a scheme. Such HTAs can be conducted for example, in the context of i) a request from a manufacturer to include the device on a positive reimbursement list (e.g., Belgium, France—PRS, the Netherlands and Switzerland); ii) a request from a provider for an extra remuneration of the procedure involving the device, for example on top of an existing diagnosis-related group -DRG tariff (e.g., in Germany); or iii) a request for an evaluation of a procedure or device already in use in clinical practice (e.g., in Belgium, Germany, Spain and Switzerland). Secondly, a device could be selected following an active screening of potential candidates for CED schemes conducted by the decision body or by a committee specifically appointed for this task (e.g., in England or Spain). Finally, a device could be selected following a direct application to initiate a CED scheme by manufacturers or other stakeholders, such as care providers and health insurers with an interest in the device (e.g., in Belgium, France—FI studies, the Netherlands and Switzerland).
In all jurisdictions criteria are used to select and/or prioritize devices for inclusion in a scheme, and decisions are made either through a deliberative process or using an explicit scoring system or checklist. However, a formal assessment of the pros and cons of initiating a scheme, as opposed to other policy decisions, such as providing unconditional coverage, or refusing to adopt the device until better evidence becomes available, was never clearly defined.
Designing a CED scheme
We identified differences in the design of schemes between countries. For example, Spain and Switzerland mainly operated schemes in which a device is reimbursed for all indicated patients while data are collected in a subset of patients (i.e., only with research—OWR), whereas England, the Netherlands and Belgium mainly operated schemes in which a device is reimbursed only for patients who enrol in a clinical study (i.e., only in research—OIR). In France schemes were either OIR in the FI programme or OWR in the PRS, whereas in Germany the type of schemes depended on whether the technology was intended for inpatient use (OWR) or outpatient use (OIR).
It is worth noting, however, that within countries, the designs were relatively similar between schemes and appeared not to be tailored to (the specific characteristics of) the device under evaluation or to key sources of uncertainty. Moreover, the study designs were similar between schemes in each country and mainly concerned either observational designs utilizing real-world data, or experimental designs to ensure a high level of evidence (e.g., randomized controlled trials).
Implementing a CED scheme
We observed differences in the governance of CED schemes as well as in the roles and responsibilities of the stakeholders involved, regarding the development of the research protocol and the subsequent monitoring of the scheme. Overall, we identified two main approaches. In the first approach (e.g., in France, the Netherlands and Switzerland), the responsibility for the development of the study protocol, the monitoring of the scheme and the quality of the generated data relies entirely on the scheme applicants (e.g., the manufacturer or care providers). However, in defining the study protocol the applicants typically must follow the recommendations of the relevant decision body. Usually, the protocol requires formal approval before study initiation, to make sure that it is suitable for addressing the identified uncertainties regarding the device. In the second approach, the responsibility for the development of the study protocol and the quality of the generated data are coordinated centrally (e.g., by HTA agencies), and managed either directly or through third-party research centres (e.g., in Belgium, England, Germany and Spain).
Patient representatives may be involved in the initial assessment phase on the desirability of the scheme (e.g., in Spain or France) or later during the recruitment phase of the study (e.g., in England), but their involvement in the design phase and the development of the protocol was generally limited.
During the scheme, the costs of care provision (including utilization of the device) are usually funded through the public health care system. Specific funding arrangements may be defined at the onset to cover the additional costs of the device or procedure, by either establishing a forfeit or negotiating an add-on to an already existing DRG tariff. However, different arrangements exist for covering the additional costs associated with the research, including the costs of developing the study protocol, scientific monitoring, data collection and analysis. These costs may be either entirely financed with public funds (e.g., in Belgium, England and Spain) or they may be partially or entirely covered by the scheme applicant (e.g., in France, Germany, the Netherlands and Switzerland). Notably, in some cases, funding arrangements also include resources for data collection. In other cases, health care providers are required to perform this task without any additional funding, for example, as a condition of participating in the scheme and gaining market access for the device (e.g., in Spain or Belgium).
Evaluating a CED scheme
Decisions at the end of schemes mainly concerned the confirmation of the reimbursement status of the device, the refinement of clinical indications or conditions of use. For most of the identified schemes, no ex-ante decision rules that explicitly linked the scheme results to future decisions were defined. In most countries the schemes solely concerned the collection of additional evidence to reduce the identified uncertainties, while the final decision on the reimbursement, coverage or use of the device was integrated in the routine decision-making framework. A notable exception was the Netherlands, where the level of effectiveness that must be demonstrated during the scheme to obtain unconditional reimbursement was predefined at the onset of the scheme, in a covenant agreement signed by all stakeholders. Moreover, the covenant also addressed how to manage the withdrawal of a device in case it proved to be insufficiently effective or the data did not allow an informed decision (e.g., due to poor data quality or inconclusive results).
Notably, all participants reported having no, or only very little experience, with schemes that led to a negative coverage decision. Indeed, of the 24 CED schemes for which information on final decisions were available, coverage was confirmed (or conditional coverage prolonged due to data quality issues) in 22 cases.
Challenges associated with CED schemes for medical devices
Of the 25 participants, 18 scored the 13 challenges on the six-point Likert scales. Of these, nine were from jurisdictions with CED programmes involving devices, and nine were from jurisdictions with CED programmes involving drugs only. The seven participants who did not score the challenges were from countries without CED programmes.
For most of the assessed challenges, scores were observed across the full range of the Likert scales, indicating no clear patterns in the decision-makers’ perceptions. Table 4 presents the mean and median scores for each challenge. Overall respondents from jurisdictions with CED programme for medical devices tended to give lower scores to most of the challenges as opposed to respondents from jurisdictions without such programmes. However, the low sample size and the variability in responses within each challenge hampered any firm conclusion.
Table 5 presents the main factors that, according to the participants, positively or negatively influenced the challenges. Many of the factors identified are common to all technologies and consistent with the existing literature on CED schemes. However, some elements specific to devices could be identified.
Devices were generally considered to be more difficult to identify and monitor than pharmaceuticals, given that their routes to market are often less clear and may not be observed by those who are responsible for selecting potential candidates for CED schemes. The intrinsic characteristics of devices were also reported to pose additional challenges in the design and implementation of schemes. For example, device-user interactions and the context-specific factors which may affect device performance in the real-world were considered as challenges for the identification of all relevant uncertainty at the time of scheme initiation, and for the definition of the study protocol. In addition, devices may be associated with uncertainties that cannot be easily resolved within a feasible time frame for a scheme, such as uncertainties over the devices’ durability or their long-term performance in patients with different clinical conditions and physiologies. This in turn may increase the tension between the need to pragmatically rely on surrogate endpoints, which are rarely validated for MD procedures, and the relevance of the data collected to inform decision-making at the end of the scheme. In addition, routinely collected data, such as administrative datasets or electronic health records were expected to be less often available, or relevant, for devices, as compared with pharmaceuticals.
Relating to the possibility of product modifications during the timeframe of the scheme, one of the main concerns related to the fact that such modifications could bias the results of the study or compromise the relevance of the new evidence collected. In this respect, being able to anticipate product modifications by means of dialogues with manufacturers and sharing of information was considered a potentially mitigating factor. However, the possibility of product modifications was not perceived by most of the respondents as a major challenge, or something which is likely to occur during the duration of a scheme.
Similarly, about half of the respondents did not consider the possibility that similar products would enter the market during the period of the scheme to be an important challenge. Possible reasons related to the fact that most of the schemes evaluate a class of devices or a procedure rather than a single branded device, or that, even if focussed on a single product, they collected mainly non-comparative data. However, other respondents emphasised the difficulty of anticipating which products would enter the market during the schemes and the possibility that relative effectiveness estimates may not be meaningful anymore by the end of the scheme, as clinical practice changes more rapidly in the context of devices compared to pharmaceuticals.
Finally, with respect to the existence of a learning curve, interviewees acknowledged it as a challenge which affects both the collection and analysis of data, as well as the design of the study, such as deciding on the number of clinical centres authorized to use the device as part of the scheme. However, direct experience with this aspect was generally limited across all respondents.
Discussion
CED schemes and their application to medical devices are important items on the policy and research agendas. The objectives of this research were to explore the characteristics and use of CED schemes for devices in Europe, as well as the challenges that decision-makers face when designing and operating these schemes. Our study importantly adds to the existing knowledge base by providing a comprehensive and multi-country overview, which was directly informed by surveys with European decision/makers.
We found that 78 device-related CED schemes have been operated over the last 5 years in European countries. However, only seven countries had CED programmes in place for medical devices. To a large extent, this result may reflect the uneven application of HTA within Europe, since it may be difficult to develop a policy for CED schemes without having an established HTA capacity. For example, deciding that more data are required post-launch implies that some form of assessment of clinical or cost-effectiveness has been made. Nevertheless, HTA capacity cannot fully explain these differences, since CED schemes seem to be less frequently used for devices than for drugs [15].
The characteristics of the identified CED programmes underpinning the individual schemes for devices varied between countries, which may reflect local differences in how HTA is organised and practised. For example, schemes were either initiated by the authorities (i.e., Ministry of Health), often as a consequence of the findings of an HTA for the technology, or as a response to a request from a manufacturer. We found similar patterns in the relative responsibilities for the funding of schemes and the design of study protocols although the authorities always played some role in study design, either by outlining a general specification or recommending that an independent research centre be involved. These differences in roles were also found in the aspects of the implementation of schemes, including the collection and analysis of data, which was sometimes the responsibility of the manufacturer and sometimes an independent party.
One aspect that deserves attention is how devices are selected for a scheme. Indeed, CED is not a costless activity and its (opportunity) costs and benefits should be considered alongside other policy options, such as adopting or refusing adoption of the technology, based on currently available data, or negotiating a lower price. Aspects to be considered should include: 1) the expected value of research option(s) in terms of reduced uncertainty; 2) the direct costs of collecting evidence; 3) the opportunity costs of any delay in providing access to the technology because of the scheme; and 4) the existence of any irreversibility in the process (e.g. difficulty to subsequently withdrawal the technology, or difficulty to conduct further research after conditional approval) [1, 13]. However, while all the identified programmes used criteria to identify and prioritize technologies for a scheme, a formal assessment of these aspects was generally missing. Related to the previous point. In many jurisdictions, there does not seem to be an option for choosing among different types of CED schemes, such as OWR and OIR schemes. Nonetheless, also depending on characteristics that are specific to, or particularly relevant for devices (e.g., the existence of irreversible upfront investment costs), there may be cases where either one or the other type of CED scheme would be optimal [13, 18]. As reported in the recent report from the ISPOR good practice Task Force, Value of Information (VOI) analysis may be used to support formal assessments on the opportunity to initiate a CED scheme and the type of scheme which maximizes optimal allocation of healthcare and research funds [19].
In addition, one general finding across all countries was that relatively little attention seemed to be paid to the evaluation of schemes, both in itinere during data collection and at the time of the reassessment of the technology once the scheme reported its results. This mirrors the findings of other studies of CED and market access schemes more generally [1, 20, 21] and is obviously an area that requires further attention by policy makers and researchers. Indeed, issues with the quality and timely reporting of data have been mentioned as a factor hampering CED schemes (see e.g., Table 5). For example, in France, where manufacturers are solely responsible for the collection of additional data, the lack of the requested evidence from post-registration studies was often reported in the technology re-appraisals.
The policy responses at the end of a CED scheme for devices may be more complicated than, for example, deciding on whether to include a drug on a formulary or to determine prescribing guidelines, since the reimbursement of devices, and the policies to determine their use, are often linked to the use of broader surgical, or other treatment, interventions. Therefore, policies probably involve adjustments to DRG tariffs, or changes to clinical guidelines, and/or hospital practice more generally. Hence, decision rules and policies for discontinuing the use of devices require attention in this context.
Notably, all participants reported to have no or little experience with refusing to confirm reimbursement at the end of the schemes. While this may reflect the degree and type of uncertainties existing at the beginning of the schemes, it may also signal a certain difficulty in reversing the preliminary reimbursement decision once a technology has entered a scheme [17, 22, 23]. This aspect may be even more relevant if no ex-ante criteria for evaluating the schemes were defined, as was the case for almost all schemes for devices in Europe.
Based on our observations of variation in the characteristics of schemes, it is difficult to prescribe a single preferred approach to CED of devices in Europe. Each country has specific local differences in HTA practices, although knowledge on how CED schemes have been used elsewhere can be used to develop local guidance. However, ideally a primary driver of the initiation of CED schemes would be the outcome of HTAs for the technologies concerned, since this can help identify the uncertainties in (cost-) effectiveness that (in principle) could be resolved through CED.
The participants’ perceptions of the various challenges in initiating, designing, implementing, and evaluating CED schemes were varied and did not indicate that, in general, some challenges were substantially more important than others. The reasons for this are unclear, although in some cases the participant’s perception of a given challenge reflected local circumstances. For example, funding was not perceived as a major challenge in settings where public funding was made available, but a major challenge in settings where it was not. In addition, the scores obtained for those challenges that were ‘device specific’ did not differ substantially from those for the other, more generic challenges. While this aspect requires further investigation, our general impression was that some of the low scores given for ‘device specific’ challenges are attributable to a lack of direct experience with addressing these issues, given that the use of CED schemes for medical devices in some European countries is generally quite recent. For example, it has been argued that manufacturers may be reluctant to engage in a scheme and generate new evidence if other competitors entering the market with fast-follower products could also benefit from it [24]. So, one option would be to require that each manufacturer generates the same clinical evidence as for devices already on the market, unless there is compelling evidence of ‘equivalence’ for the new device [24, 25]. However, this option risks a waste of (public) resources in conducting clinical studies that are not strictly necessary. Moreover, the consequences of such a strategy in terms of competitiveness, market prices and eventually access of potentially valuable devices to patients remain largely unexplored.
We observed that the scores for the challenges were lower for respondents in countries where there was direct experience in CED for devices, as compared with those having experience with CED for drugs only. However, although the numerical differences in the scores were substantial, the small sample size means that no firm conclusions can be drawn. This could be explored in further research by comparing decision-makers’ perceptions before and after operating CED schemes and relating these perceptions to the general (HTA) infrastructure in a country.
We used a combination of methods to obtain insights in the use of and challenges related to CED schemes in the relatively understudied context of devices, including a large set of European countries. The insights obtained allow learning from experiences across countries and increase the chances of having successful CED schemes in the future, by highlighting how decision makers perceive and deal with specific challenges. Nonetheless, some limitations also need highlighting. First, although we studied experiences in many European countries, we cannot be sure that our overview is complete as some countries were not included in the study. Moreover, although in each country we interviewed the person we considered to be most knowledgeable about CED schemes, we cannot be sure that the views of the participants are representative of the views of decision-makers more generally. Additionally, we focussed on the detailed perceptions of decision-makers, with a focus on HTA agencies at the national or regional level and (some) national payers because recent research suggests that decision-makers may be hesitant to engage in CED schemes [5]. This makes them not only a relevant source for the current study in terms of knowledge, but also in articulating (potential) challenges and difficulties with applying such schemes. Future studies could nonetheless supplement this with information on the perceptions of other stakeholders, such as clinical professionals, patient organisations, local payers/decision makers, and manufacturers. Finally, our focus was on schemes initiated at the national or regional level. In addition, some schemes involving devices may be negotiated at the local level directly between providers and manufacturers. Many of these may be ‘pay for performance’ schemes, but some could be characterized as CED schemes. These schemes were outside the scope of our current study, but their characteristics and performance are nonetheless important to investigate further.
Conclusions
CED schemes for medical devices offer a promising tool to increase value for money in health care. While they are currently used in Europe, this study has shown experience with these schemes to be limited to a relatively small number of countries. Moreover, considerable variation exists between countries in how schemes are initiated, designed, implemented, and evaluated.
While the identified challenges in using CED schemes were perceived differently, none of them was unanimously considered insignificant. Hence, all challenges should be considered when initiating CED schemes in a given country. Our recommendation is that each jurisdiction embarking on CED schemes for devices should undertake its own ‘risk assessment’, using our list of challenges as a starting point, and considering for each of them the factors that decision-makers in this study outlined as having either a positive or negative influence. If a given challenge is considered to be important locally, the highlighted experiences of other countries in this study can help in addressing or overcoming them. That way, this study directly contributes to making CED schemes for devices a more effective policy option in the future.
Availability of data and material
The full database with information on the 78 CED schemes collected is provided in Online Resource 4.
References
Garrison, L.P., Towse, A., Briggs, A., de Pouvourville, G., Grueger, J., Mohr, P.E., et al.: Performance-based risk-sharing arrangements—good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health 16(5), 703–719 (2013)
Levin, L., Goeree, R., Levine, M., Krahn, M., Easty, T., Brown, A., et al.: Coverage with evidence development: the Ontario experience. Int. J. Technol. Assess. Health Care 27(2), 159–168 (2011)
Tunis, S.R., Pearson, S.D.: Coverage options for promising technologies: medicare’s ‘coverage with evidence development.’ Health Aff. 25(5), 1218–1230 (2006)
Dabbous, M., Chachoua, L., Caban, A., Toumi, M.: Managed entry agreements: policy analysis from the European perspective. Value Health 23(4), 425–433 (2020)
Schaffer, S.K., Messner, D., Mestre-Ferrandiz, J., Tambor, E., Towse, A.: Paying for cures: perspectives on solutions to the “Affordability Issue.” Value Health 21(3), 276–279 (2018)
Faulkner, E., Spinner, D.S., Ringo, M., Carroll, M.: Are global health systems ready for transformative therapies? Value Health 22(6), 627–641 (2019)
Carlson, J.J., Chen, S., Garrison, L.P.: Performance-based risk-sharing arrangements: an updated international review. Pharmacoeconomics 35(10), 1063–1072 (2017)
Piatkiewicz, T.J., Traulsen, J.M., Holm-Larsen, T.: Risk-sharing agreements in the EU: a systematic review of major trends. PharmacoEconomics Open 2(2), 109–123 (2018)
Pauwels, K., Huys, I., Vogler, S., Casteels, M., Simoens, S.: Managed entry agreements for oncology drugs: lessons from the european experience to inform the future. Front. Pharmacol. [Internet] (2017). https://doi.org/10.3389/fphar.2017.00171/full. (accessed 22 May 2020)
Serrano-Aguilar, P., Gutierrez-Ibarluzea, I., Díaz, P., Imaz-Iglesia, I., González-Enríquez, J., Castro, J.L., et al.: Postlaunch evidence-generation studies for medical devices in Spain: the RedETS approach to integrate real-world evidence into decision making. Int. J. Technol. Assess. Health Care 37(1), e63 (2021)
Tarricone, R., Ciani, O., Torbica, A., Brouwer, W., Chaloutsos, G., Drummond, M.F., et al.: Lifecycle evidence requirements for high-risk implantable medical devices: a European perspective. Expert Rev. Med. Devices 23, 1–14 (2020)
Drummond, M., Griffin, A., Tarricone, R.: Economic evaluation for devices and drugs—same or different? Value Health 12(4), 402–404 (2009)
Rothery, C., Claxton, K., Palmer, S., Epstein, D., Tarricone, R., Sculpher, M.: Characterising uncertainty in the assessment of medical devices and determining future research needs: characterising uncertainty in the assessment of devices. Health Econ. 26, 109–123 (2017)
Medtech Europe. The European Medical Technology Industry – in figures 2019 [Internet]. (2019) Available from: https://www.medtecheurope.org/wp-content/uploads/2019/04/The-European-Medical-Technology-Industry-in-figures-2019-1.pdf. Accessed 10 May 2021
Medtech Europe. Taxonomy of Value-Based Access Programmes—Funding for Innovation [Internet]. Medtech Europe. (2020) Available from: https://assets.medtechweek.eu/Hot-Topics-2019-documents/Taxonomy%20of%20Value-Based%20Access%20Programs-Guidance-Doc.pdf. Accessed 10 May 2021
Medtech Reimbursement consulting. Innovative payment schemes for medical technologies and invitro diagnostic tests in Europe—MTRC research paper [Internet]. European Med Tech and IVD Reimbursement Consulting. (2018) Available from: https://www.medtecheurope.org/wp-content/uploads/2018/06/2018_MTE_MTRC-Research-Paper-Innovative-Payment-Schemes-in-Europe.PDF. Accessed 10 May 2021
Reckers-Droog, V., et al.: Challenges with coverage with evidence development schemes for medical devices: a systematic review. Health Pol Technol 9(2)146–156 (2020)
Grutters, J.P.C., Abrams, K.R., Ruysscher, D., Pijls-Johannesma, M., Peters, H.J.M., Beutner, E., et al.: When to wait for more evidence? Real options analysis in proton therapy. Oncologist 16(12), 1752–1761 (2011)
Fenwick, E., Steuten, L., Knies, S., Ghabri, S., Basu, A., Murray, J.F., et al.: Value of information analysis for research decisions—an introduction: report 1 of the ISPOR value of information analysis emerging good practices task force. Value Health 23(2), 139–150 (2020)
Gerkens. S., Neyt, M., San Miguel, L., Vinck, I., Thiry, N., Cleemput, I.: How to improve the belgian process for managed entry agreements? An analysis of the belgian and international experience [Internet]. Belgium KCE. (2020) Available from: https://kce.fgov.be/sites/default/files/atoms/files/KCE_288_Improve_Belgian_process_managed_entry_agreements_Report.pdf. Accessed 10 May 2021
Kanavos, P., Ferrario, A., Tafuri, G., Siviero, P.: Managing risk and uncertainty in health technology introduction: the role of managed entry agreements. Glob. Policy 8, 84–92 (2017)
Stafinski, T., McCabe, C.J., Menon, D.: Funding the unfundable: mechanisms for managing uncertainty in decisions on the introduction of new and innovative technologies into healthcare systems. Pharmacoeconomics 28(2), 113–142 (2010)
van de Wetering, E.J., van Exel, J., Brouwer, W.B.F.: The challenge of conditional reimbursement: stopping reimbursement can be more difficult than not starting in the first place! Value Health 20(1), 118–125 (2017)
Brugger, U., Horisberger, B., Ruckstuhl, A., Plessow, R., Eichler, K., Gratwohl, A.: Health technology assessment in Switzerland: a descriptive analysis of ‘Coverage with Evidence Development’ decisions from 1996 to 2013. BMJ Open 5(3), e007021–e007021 (2015)
Drummond, M., Tarricone, R., Torbica, A.: Incentivizing research into the effectiveness of medical devices. Eur. J. Health Econ. 17(9), 1055–1058 (2016)
Acknowledgements
The authors thank all COMED project members, and particularly the project leader Aleksandra Torbica, for their helpful comments on an earlier version of this paper. The authors also want to thank the participants to the survey for their thoughtful answers during the interview and their continuous support following any further request for clarification and/or validation.
Funding
Open access funding provided by Università Commerciale Luigi Bocconi within the CRUI-CARE Agreement. This study has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 779306 for the project “Pushing the boundaries of cost and outcome analysis of medical technologies—COMED”.
Author information
Authors and Affiliations
Contributions
Conceptualization: CF, VR-D, WB, MD; Methodology: CF, VR-D; Formal analysis and investigation: CF; Writing—original draft preparation: FC, VR-D, MD; Writing—review and editing: OC, FD, BG, ZK, SK, KS, WB, MD; Supervision: WB, MD.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study is part of the EU Horizon 2020 COMED project that has been reviewed and approved by the Bocconi University Ethics Committee (protocol number: 0068538, approved on May 8, 2018).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Federici, C., Reckers-Droog, V., Ciani, O. et al. Coverage with evidence development schemes for medical devices in Europe: characteristics and challenges. Eur J Health Econ 22, 1253–1273 (2021). https://doi.org/10.1007/s10198-021-01334-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10198-021-01334-9
Keywords
- Coverage with evidence development
- Medical devices
- European HTA policies
- Value of information
- Adoption and reimbursement of medical devices