
Abstract
Background
EOX (epirubicin, oxaliplatin, and capecitabine) is one of the standard regimens for metastatic or locally advanced gastric cancer (GC). A new combination based on fractional docetaxel (low-TOX) has been developed in an attempt to increase the efficacy of EOX and reduce the heavy toxicity of classical docetaxel regimens.
Methods
Overall, 169 previously untreated GC patients were randomized between EOX (arm A) and low-TOX (arm B). The primary endpoint was progression-free survival (PFS), while secondary ones were overall survival (OS), overall response rate (ORR), disease control rate (DCR), and tolerability. The study was designed to detect a 35% (80% power at a two-sided 5% significance level) PFS increase with low-TOX and an interim analysis for futility was planned after the first 127 events.
Results
At the cut-off date of interim analysis, median PFS was 6.3 months [95% confidence interval (CI) 5.0–8.1] in arm A vs 6.3 months (95% CI 5.0–7.8) in arm B, without statistical difference. OS was comparable in the two arms: 12.4 in arm A (95% CI 9.1–19.2) vs 11.5 months in arm B (95% CI 8.6–15.0). ORR was 33% and 24%, while DCR was 68% and 67%, respectively. Treatment modification (91% vs 78%, P = 0.017) and number of patients with CTC grade ≥ 3 adverse events (42 vs 35) were higher in arm B.
Conclusions
A triplet regimen based on the fractional dose of docetaxel achieves no improvement over EOX which remains a potential standard treatment in many patients with inoperable, locally advanced or metastatic GC.
Similar content being viewed by others


Introduction
Although the incidence of adenocarcinoma of the stomach is slowly decreasing, over 26,000 new cases are estimated in the United States with over 11,000 deaths expected in 2021 alone [1].
In patients with advanced disease, chemotherapy improves overall survival (OS) in comparison to best supportive care [2, 3]. Combinations of two or three drugs including a platin derivative (cisplatin or oxaliplatin), a fluoropyrimidine [5-fluorouracil (5-FU) or oral capecitabine], and often an anthracycline (usually epirubicin) have demonstrated superiority compared to single agent therapy and are the current standard [4]. REAL-2 is so far the largest study (about 1000 patients) evaluating several triple combinations of these agents (ECF, ECX, EOF, and EOX) [5]. The study proved that oxaliplatin was better tolerated than cisplatin and that the substitution of 5-FU with capecitabine did not decrease survival. All four combinations were equivalent for OS (median 9.3–11.2 months) and for progression-free survival (PFS) (median 6.2–7.0 months) with acceptable toxicity. However, EOX showed longer OS than ECF (HR 0.80, median 9.9 months vs 11.2 months) and has become a preferred reference regimen in ongoing phase III studies.
Docetaxel is another active agent that, when added to cisplatin and 5-FU, has shown to significantly improve OS (9.2 months vs 8.6 months), time to progression (TTP) (5.6 months vs 3.7 months), and response rate (RR) (35% vs 24%) [6]. Moreover, patients receiving this triplet regimen (DCF) had better preservation of quality of life compared to those treated with the doublet combination (cisplatin plus 5-FU), although DCF showed increased toxicity [7]. Several investigators have attempted to modulate DCF toxicity mostly within phase I studies by replacing cisplatin with oxaliplatin and 5-FU with capecitabine and varying the dose of the three agents [8,9,10,11,12]. As of today, there are no published studies comparing anthracycline-based to taxane-based three-drug regimens.
The aim of the LEGA trial is to compare the EOX regimen evaluated in the REAL-2 study in patients with HER2 negative tumors with another three-drug regimen containing docetaxel, oxaliplatin, and capecitabine whose dosages and weekly taxane schedule were conceived based on the aforementioned phase I studies.
Patients and methods
Study design
The LEGA trial is a randomized, parallel group, non-blinded phase III trial comprising patients with advanced (loco-regional or metastatic) HER2 negative or unknown tumor who had not been previously treated with chemotherapy for this stage. Patient enrollment was carried out in 23 oncology centers. This is an investigator-initiated trial; therefore, the sponsor is Fondazione GISCAD (Gruppo Italiano per lo Studio dei Carcinomi dell'Apparato Digerente) and the Medical Oncology Department—Azienda Ospedaliera Ospedali Riuniti of Bergamo acted as the coordinator site. The study was approved by ethical committees of each participating center and conducted according to the Declaration of Helsinki and Good Clinical Practice guidelines. The trial is registered with ClinicalTrials.gov, registration number: 2011-005537-39.
Participants
Eligible patients had histopathological proven metastatic or locally advanced, non-resectable gastric adenocarcinoma with Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–1. Participants had received no prior chemotherapy except for adjuvant therapy completed at least 1 year before study entry and had a life expectancy of at least 3 months. Additionally, hematological, liver, and renal functions had to be within normal range; neutrophils ≥ 2.0 × 109/L, platelets ≥ 100 × 109/L, and hemoglobin ≥ 10 g/dL, bilirubin level either normal or ≤ 1.5 × UNL, serum creatinine < 1.5 × ULN. In the presence of borderline values, the calculated creatinine clearance should have been ≥ 60 mL/min. Exclusion criteria included evidence of CNS metastasis; concurrent chronic systemic immune therapy; clinically relevant coronary artery disease or a history of a myocardial infarction or a history of hypertension not controlled by therapy within the last 12 months; known hypersensitivity to study drugs; known grade 3 or 4 allergic reaction to any of the components of the treatment; known drug abuse/alcohol abuse; acute or subacute intestinal occlusion and any other significant chronic gastrointestinal disease that could interfere with absorption of oral treatment; history of clinically relevant psychiatric disability precluding informed consent; presence of any psychological, familial, sociological or geographical condition potentially hampering compliance with the study protocol and follow-up schedule; pregnant or breastfeeding women; active uncontrolled infection(s); positivity for HIV serology and/or viral hepatitis B or C; any concurrent malignancy other than non-melanoma skin cancer, or carcinoma in situ of the cervix. All patients provided written informed consent before enrollment.
Randomization
Upon completion of screening evaluation, the patient, if eligible, was randomized via a web-based application made available to the centers. The following information was collected at the time of randomization: site identification number, patient’s date of birth, gender, and ECOG PS. Patients were treated according to a 1:1 ratio and they were stratified by PS, 0 or 1. The time between randomization and initiation of treatment could not be longer than 7 days. Patients were assigned to receive either epirubicin 50 mg/m2 intravenously (iv) day 1 + oxaliplatin 130 mg/m2 iv day 1 + capecitabine 625 mg/m2 orally b.i.d. days 1–14 (EOX—arm A) or docetaxel 35 mg/m2 iv days 1 and 8 + oxaliplatin 80 mg/m2 iv day 1 + capecitabine 750 mg/m2 orally b.i.d. days 1–14 (low-TOX—arm B). Treatment cycles of both arm A and B were repeated every 3 weeks.
Procedures
Toxicity was evaluated after each chemotherapy cycle and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0. Treatment was interrupted in cases of grade 2 toxicity or worse and was resumed once toxicity improved to grade 0 or 1. In any case, treatment was delayed for no more than 2 weeks. Dose modification criteria were predefined, based on the worst grade of an adverse event (AE) or laboratory abnormality, and performed in case of severe hematological (neutropenia and thrombocytopenia grade 4) or gastrointestinal (stomatitis and diarrhea grade 3–4) toxicity. If multiple adverse events were observed, the dose administered was based on the most severe (worst grade) event.
In these cases, the drugs were then restarted as follows: epirubicin 40 mg/m2 intravenously (iv) day 1 + oxaliplatin 80 mg/m2 iv day 1 + capecitabine 300 mg/m2 orally b.i.d. days 1–14 (EOX—arm A) or docetaxel 25 mg/m2 iv days 1 and 8 + oxaliplatin 50 mg/m2 iv day 1 + capecitabine 350 mg/m2 orally b.i.d. days 1–14 (low-TOX—arm B), with cycles repeated every 3 weeks. Treatment was discontinued in cases of further grade 3–4 toxicity. If the patients stopped treatment for > 2 weeks for any reason other than side effects, they were withdrawn from the trial for non-compliance.
Eligible patients remained on therapy for a maximum of 6 cycles unless clinical or radiological progression or occurrence of other events causing withdrawal took place and had their tumor status assessed according to RECIST 1.0 criteria [13] within 28 days prior to initiation of treatment and to all 3 cycles. After documented disease progression (PD), information on survival status and post-study chemotherapy onco-specific treatments was collected in all patients every 12 weeks up to 18 months from date randomization. Non-progressing patients withdrawn before completion of the planned number of cycles underwent tumor assessment within 4 weeks from treatment discontinuation (unless a tumor assessment had been performed in the 12 weeks preceding the withdrawal); thereafter, they were monitored in a similar way as patients completing treatment.
Physical examination was performed at least 7 days prior to the initiation of the study treatment and within 28 days from treatment discontinuation. Temperature, weight, height, and other safety indicators [adverse events (AEs), laboratory tests, etc.] were collected prior to the initiation of each treatment cycle. In arm B hematologic tests were repeated also before the administration of docetaxel on day 8. Electrocardiogram (ECG) and left ventricular ejection fraction (LVEF) evaluation with echocardiogram or multigated acquisition (MUGA) were performed on all patients at baseline and LVEF was repeated at least 28 days after treatment discontinuation to patients treated with EOX, unless clinically indicated before.
Outcomes
The primary endpoint was PFS, calculated from the date of randomization to the first date of radiological or clinical progression on first-line chemotherapy, or death by any cause, whichever came first. Subjects who had not progressed or died during the study or were lost to follow-up were censored at the last disease assessment performed within the cut-off date. Secondary endpoints were OS, defined as time from the date of randomization until death by any cause (living subjects were censored at cut-off date); overall response rate (ORR); disease control rate (DCR), defined as the sum of patients achieving an evaluable complete radiological response (CR), partial response (PR), or stable disease (SD); toxicity [14].
Statistical considerations
Based on published results, the expected median PFS of patients receiving EOX is estimated to be around 7 months [5]. The current study was designed to test whether low-TOX regimen could provide a 35% reduction of the hazard of progression or death as compared to EOX, meaning an improvement in median PFS for up to about 11 months [i.e., hazard ratio (HR) ≤ 0.65 under alternative hypothesis]. In this case, the experimental drug combination would be considered effective.
To detect such an improvement with 80% power at a two-sided 5% significance level, the required overall sample size was 190 enrolled subjects. An interim analysis was planned to consider whether the trial should be stopped for futility. Such analysis was planned to be conducted after the first 127 events (75% of the total number). Conditional Power (CP) method was applied for the current study [15]. The threshold was set to 30% and the trial could be stopped early if the computed CP was below this value.
All data analyses were performed after the database had been released and as indicated at the beginning in the statistical analysis plan that considered any amendment to the protocol. Statistical programming and analyses were carried out using validated statistical software (SAS 9.4).
Descriptive statistics were used to report patient disposition and demographic variables in all the enrolled and randomized patients. Distribution of these data was presented by summary statistics such as median, minimum and maximum, mean and standard deviation for quantitative outcomes; frequency distributions were used for the categorical/categorized variables. The same method was applied to describe treatment administration (e.g., number of cycles, treatment delays/modifications), safety analysis (e.g., number/percentage of patients with adverse events) and laboratory assessments on the treated patient population.
Survival curves of the two arms were compared by the log-rank test stratified by PS (0 or 1) and the Kaplan–Meier (KM) method was used to estimate cumulative survival probability [16]. Likewise for PFS, the KM method was applied for OS. Point estimates and 95% confidence interval (CI) estimates were calculated for ORR, as well as for disease control rate (DCR). The between treatment comparison was performed by Mantel Haenszel chi–squared test [17], controlling for ECOG PS.
Results
From January 21, 2013, to May 14, 2018, 169 patients were randomly enrolled in 23 Italian centers. Data collected recorded a total number of 164 subjects known to be treated, 82 for each arm. Five patients randomized in the control arm had never been treated and went off-study due to investigator’s decision (one patient), consent withdrawal (two patients), and early death (two patients).
Overall, among the whole treated population, 71 patients (43%) completed treatment as per protocol. The major reasons for treatment discontinuation were PD in 75 patients (46%), physician’s decision (15 patients, 9%), and toxicity (14 patients, 8%), while the death event was reported for 4 patients (2%). Patients declared as off-study amounted to 139 randomized subjects. Among these patients, the main reason was due to death (116 enrolled patients, 69%). An overview of the disposition of subjects, by group, is outlined in Fig. 1.

Trial profile
Table 1 reports the tumor characteristic frequency distribution for both study groups, as well as for the whole randomized population. The median age was 62 years (range 31–84), 65% of patients were male, and ECOG PS score was 0 in 128 of them (76%), and 1 in 37 cases (22%). As expected, no relevant differences emerged between the two groups for these factors.
On April 17, 2019, cut-off date, all the 164 treated patients had discontinued treatment. For each of the two study groups, almost half of the subjects completed treatment as per protocol (41% in low-TOX vs 45% in EOX). The most common reasons for early discontinuation in both groups was PD (37 subjects in arm A and 38 in arm B) and toxicity (14 patients). The treated patients received a total of 273 cycles. The median duration of treatment was 18.2 weeks (range, 3.0 to 30.7) in the experimental arm and 18.0 weeks (range, 3.0 to 25.1) in the EOX group. Treatment modification occurred for most of the treated patients (91% low-TOX vs 78% in EOX, P = 0.017) and was mainly caused by general disorders (P = 0.032), gastrointestinal (P = 0.010) and skin (P = 0.004) toxicities of the experimental arm, but also by poor treatment compliance. Cycle delays occurred in 48 patients (58%) for EOX and 59 patients (72%) for low-TOX. Dose reductions occurred in 29 patients with EOX (35%) and 41 patients with low-TOX (50%). Dose omissions were more frequent in arm B than in arm A (54% vs 34%, P = 0.012). The most common AE leading to cycle delay was diarrhea for low-TOX and neutropenia for EOX. A summary of the frequency distribution of treatment modification is reported in Table 2. While the median actual dose intensities of oxaliplatin and capecitabine were similar in both arms, that of docetaxel and epirubicin was 79% in arm B and 90% in arm A, respectively. The dose intensity of each drug, expressed in mg/m2/week for both arms, is shown in the Supplementary Table.
Overall, 52 patients (63%) received chemotherapy as second- or third-line in arm A, prevalently taxane-based with or without ramucirumab. Irinotecan-based chemotherapy or ramucirumab was mostly prescribed as second- or third-line in 46 patients (56%) after PD in arm B.
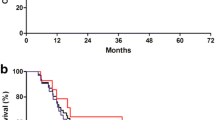
According to the protocol, an interim analysis was planned after detection of the first 127 evaluable events for the primary endpoint (PFS). This analysis showed an evident lack of improvement in favor of the experimental arm, both for primary and secondary efficacy endpoints. This assessment was also carried out by observing the calculated conditional power, which was always well below the cut-off value of 30%. These results indicated that it was unlikely that the low-TOX regimen was able to reach the target of improvement against EOX, and for this reason the decision was made to prematurely stop the study for futility. In accordance with the planned interim analysis to be performed by protocol, the data available at the cut-off date of 17 April 2019 revealed no signs of clinical evidence for the investigational regimen. Seventy events were detected in the low-TOX arm (38 were PD), whereas the number observed in the EOX arm was 62, of which 37 PD. Median PFS was comparable in the two arms (6.3 months vs 6.3 months; HR in the experimental group, 0.975; 95% CI, 0.686 to 1.384; P = 0.885). The calculated conditional power was 0.00%, which confirmed at this step the lack of possibility of reaching the 35% risk reduction target. Considering this obvious lack of evidence, no multivariate exploratory analysis was considered. The summary results are shown in Table 3 and in Fig. 2.

Kaplan–Meier curve for progression-free survival
Death occurred in 114 patients (59 in arm A vs 55 in arm B). The median OS time (Fig. 3) did not differ significantly between the low-TOX group and EOX group (11.5 and 12.4 months, respectively; HR 1.002; 95% CI 0.691 to 1.452; P = 0.992). The estimated rate of OS at 12 months was 50% in the experimental group and 51% in the control group.

Kaplan–Meier curve for overall survival
In the low-TOX group, 2 patients (2%) had a CR, 18 patients (22%) had a PR, and 35 (43%) had a stable disease (SD), whereas in the EOX group, 4 patients (5%) had a CR, 23 subjects (28%) had a PR, and 29 (35%) had a SD. The ORR rate (i.e., confirmed CR and PR) was comparable between the two arms (24% vs 33%, P = 0.585). The same conclusion was obtained for the DCR (67% vs 68%, P = 0.279).
A total of 164 patients were treated and evaluated for safety (82 per arm). Seventy-one patients completed the planned treatment cycles. Among the whole treated population, 149 subjects experienced at least one treatment emergent AE in the first or subsequent cycles. The overall incidence was 91% in the low-TOX group and 90% in the EOX group (Table 4).
No significant differences were observed between the two arms in terms of number of patients who experienced any AE, except for palmar–plantar erythrodysesthesia (27% vs 11%, P = 0.010) and diarrhea (49% vs 29%, P = 0.010) more frequent in arm B, while only neutropenia was more represented in arm A (38% vs 23%, P = 0.042). However, fatigue, nausea, vomiting, abdominal pain, erythema (15% vs 1%), rash (11% vs 1%), conjunctivitis (10% vs none), and anorexia (19% vs 7%) occurred at a higher frequency in the experimental group than in the control arm. Only hematological toxicities such as anemia, leukopenia, and thrombocytopenia were more frequently recorded in arm A. Limited to grade > 3 toxicities, diarrhea (P = 0.016) and mucositis (P = 0.013) were statistically more frequent in the experimental arm, while only neutropenia (P = 0.017) occurred more often in arm A than in arm B. Two patients died during the period of the study for reasons other than PD, one due to digestive bleeding in arm A and another due to worsening clinical conditions in arm B.
Discussion
This study had a dual purpose: to capture the potential benefits of an investigational three-drug chemotherapy regimen including docetaxel administered with weekly fractional dosage to limit potential burdensome toxicities and to compare the results with those of another historical regimen such as EOX highlighting that there is a lack of randomized head-to-head clinical trials between these triplets in the literature. The planned interim analysis was conducted when 127 patients were evaluable for the primary endpoint: patients in both groups had a median PFS of 6.3 months. Since it had been hypothesized that low-TOX regimen could provide a 35% reduction of the hazard of progression or death compared to EOX, the trial was, therefore, stopped for futility and the main endpoint was not met. The lack of efficacy improvement of low-TOX vs EOX regimen was confirmed also by evaluating the median OS (11.5 months vs 12.4 months, respectively). Other secondary endpoints such as ORR (24% vs 33%) and DCR (67% vs 68%) supported this conclusion.
Some limitations of this work should be acknowledged. The LEGA study was conceived in 2011 and, therefore, the significance and importance of our results are undoubtedly counterbalanced by the data reported with the association of chemotherapy with immunotherapy in a recent phase III trial [18]. CheckMate 649 met its dual primary endpoints of OS and PFS in patients whose tumors expressed programmed death-ligand 1 (PD-L1) with a combined positive score (CPS) of 5 or more. Based on this trial, in April 2021, the U.S. Food and Drug Administration granted approval of first-line nivolumab for patients with HER2 negative advanced GC. However, testing PD-L1 may not be a simple procedure due to its heterogeneity [19] and, although a positivity of the results was found, albeit in a more attenuated way, even in patients with CPS score < 5, the benefits of OS and PFS could have been influenced by the relatively high proportion of tumors with a CPS of 5 or more (approximately 60%) within the overall trial population [20]. Nor can the onset of side effects be ignored, such as itching, diarrhea, rash, colitis, and pneumonia which, although often manageable, could hinder the use and continuation of immunotherapy [21].
On the other hand, even though it is not clearly defined that a three-drug chemotherapy regimen can achieve a significant survival advantage over another two-drug and about thirty studies and two meta-analyses have not clarified this dilemma [22,23,24], it is important to underline how a significant increase in ORR and PFS, ensured by the former, could be useful in the most symptomatic patients and with a "bulky" disease or in potentially operable ones. Regarding the controversial use of anthracyclines, there are no randomized studies comparing regimens such as EOX with chemotherapy doublets containing platinum salts [23, 25]. So, while Wagner et al. reported a significant HR of 0.77 (95% CI 0.62–0.95) with the addition of an anthracycline based on an analysis of three randomized trials [2], other authors have pointed out how the addition of anthracycline to a doublet showed a non-significant HR of 0.7 and that the number of included patients did not reach 200 in total [24]. From this point of view, it, therefore, seemed justified to compare the two triplets in our study.
The lack of benefit of the experimental regimen compared to the one containing anthracycline could be explained by the increased toxicity of the former, although only diarrhea (P = 0.016) and mucositis (P = 0.013) of grade > 3, more frequent in arm B than in arm A, assume a statistically relevant difference. In fact, although the median duration of treatment (18.2 weeks in the experimental arm and 18.0 weeks in the EOX group) and the number of subjects who completed treatment as per protocol (41% in low-TOX vs 45% in EOX) was similar, the AEs, particularly general disorders, gastrointestinal and skin toxicities, caused treatment modifications more frequently in arm B than in arm A (P = 0.017) at the expense of a lower dose-intensity of docetaxel compared to epirubicin. As a result, more patients in the experimental arm underwent dose reductions, delays, or omissions of therapy (P = 0.012).
In this regard, it has already been shown that docetaxel-based triplets are potentially toxic, particularly due to the frequent observation of severe neutropenia and diarrhea, so they deserve careful selection, education, monitoring, and active patient management with prudential recourse to a primary prophylactic granulocyte colony-stimulating factor administration [6]. In our experience, in line with what has been demonstrated in the literature [8,9,10,11,12], although the fractional weekly administration of docetaxel confirmed a reduction of grade 3–4 toxicities compared to those reported by the DCF regimen in the Van Cutsem et al. phase III study [6], this is evidently not reflected in a benefit to the patient if we look at ORR, PFS and OS with the low-TOX regimen.
Despite being considered obsolete, the EOX regimen has been demonstrated to be safe and efficacious even in a population with unfavorable prognosis: almost 92% of the patients enrolled in this arm had metastatic disease, 64% had a poorly differentiated tumor, and 20% had a PS equal to 1. In addition, although our study also included patients with locally advanced disease (11%), they remained unresectable even after treatment, which confirms their poorer prognosis. This contrasts with other randomized trials in which the percentage of resected patients ranges from 13 to 40% after achieving a tumor response [26,27,28].
Conclusion
The final results of the LEGA trial show that replacing an anthracycline with docetaxel in a regimen that also includes oxaliplatin and capecitabine does not bring therapeutic benefits and that EOX could still be considered a regimen of choice in many patients with metastatic or inoperable locally advanced GC.
References
Siegel RL, Miller KD, Fuchs HF, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol. 2006;24:2903–9.
Wagner AD, Lx Syn N, Moehler M, Grothe W, Yong WP, Tai B-C, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2017;8:4064.
Kim JG, Chung HY, Yu W. Recent advances in chemotherapy for advanced gastric cancer. World J Gastrointest Oncol. 2010;2:287–94.
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36–46.
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 study group. J Clin Oncol. 2006;24:4991–7.
Ajani JA, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Quality of life with docetaxel plus cisplatin and fluorouracil compared with cisplatin and fluorouracil from a phase III trial for advanced gastric or gastroesophageal adenocarcinoma: the V-325 Study Group. J Clin Oncol. 2007;25:3210–6.
Andersen M, Schønnemann KR, Yilmaz M, Jensen HA, Vestermark LW, Pfeiffer P. Phase I study of docetaxel, oxaliplatin and capecitabine (TEX) as first line therapy to patients with advanced gastro-oesophageal cancer. Acta Oncol. 2010;49:1246–52.
Sym SJ, Ryu MH, Kang HJ, Lee SS, Chang HM, Lee JL, et al. Phase I study of 3-weekly docetaxel, capecitabine and oxaliplatin combination chemotherapy in patients with previously untreated advanced gastric cancer. Cancer Chemother Pharmacol. 2010;66:373–80.
Malik I, Bernal P, Byrd J. A phase I study of docetaxel, oxaliplatin, & capecitabine (DOC) as first-line therapy of patients with locally advanced or metastatic adenocarcinoma of stomach and GE junction. Cancer Invest. 2010;28:833–8.
Goel G, Jauhri M, Negi A, Aggarwal S. Feasibility study of docetaxel, oxaliplatin and capecitabine combination regimen in advanced gastric or gastroesophageal adenocarcinoma. Hematol Oncol Stem Cell Ther. 2010;3:55–9.
Grothe W, Hofheinz RD, Mantovani Loeffler L, Böhme J, Arnold D, Radestock A, et al. Phase I trial of docetaxel, oxaliplatin and capecitabine (TEX) in patients with metastatic gastric cancer. J Clin Oncol. 2006;24(18):14051.
Therasse P, Arbuck S, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–16.
Allegra C, Blanke C, Buyse M, et al. End points in advanced colon cancer clinical trials: a review and proposal. J Clin Oncol. 2007;25:3572–5.
Sully B, Julious SA, Nicholl J. An investigation of the impact of futility analysis in publicly funded trials. Trials. 2014;15:61.
Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81.
Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–48.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40.
Zhou KI, Peterson B, Serritella A, Reizine N, Wang Y, Catenacci DVT. Evaluation of spatiotemporal heterogeneity of tumor mutational burden (TMB) in gastroesophageal adenocarcinoma (GEA) at baseline diagnosis and after chemotherapy. J Clin Oncol. 2020;38(suppl):4546.
Fong C, Cunningham D. Chemotherapy with nivolumab in advanced gastro-oesophageal adenocarcinoma. Lancet. 2021;398:2–3.
Weber JS, Postow M, Lao CD, Schadendorf D. Management of adverse events following treatment with anti-programmed death-1 agents. Oncologist. 2016;21:1230–40.
Guo X, Zhao F, Ma X, Shen G, Ren D, Zheng F, et al. A comparison between triplet and doublet chemotherapy in improving the survival of patients with advanced gastric cancer: a systematic review and meta-analysis. BMC Cancer. 2019;19:1125.
Ter Veer E, Mohammad HN, van Valkenhoef G, Ngai LL, Mali RMA, Anderegg MC, et al. The efficacy and safety of first-line chemotherapy in advanced esophagogastric cancer: a network meta-analysis. J Natl Cancer Inst. 2016;108:10.
Mohammad NH, ter Veer E, Ngai L, Mali R, van Oijen MGH, van Laarhoven HWM. Optimal first-line chemotherapeutic treatment in patients with locally advanced or metastatic esophagogastric carcinoma: triplet versus doublet chemotherapy: a systematic literature review and meta-analysis. Cancer Metastasis Rev. 2015;34:429–41.
Lordick F, Lorenzen S, Yamada Y, Ilson D. Optimal chemotherapy for advanced gastric cancer: is there a global consensus? Gastric Cancer. 2014;17:213–25.
Ohtsu A, Shimada Y, Shirao K, Boku N, Hyodo I, Saito H, et al. Randomized phase III trial of fluorouracil alone v fluorouracil plus cisplatin v uracil and tegafur plus mitomycin in patients with unresectable, advanced gastric cancer: The Japan Clinical Oncology Group Study (JCOG9205). J Clin Oncol. 2003;21:54–9.
Vanhoefer U, Rougier P, Wilke H, Ducreux MP, Lacave AJ, Van Cutsem E, et al. Final results of a randomized phase III trial of sequential high-dose methotrexate, fluorouracil, and doxorubicin v etoposide, leucovorin, and fluorouracil v infusional fluorouracil and cisplatin in advanced gastric cancer: A trial of the European Organization for Research and Treatment of Cancer Gastrointestinal Tract Cancer Cooperative Group. J Clin Oncol. 2000;18:2648–57.
Webb A, Cunningham D, Scarffe JH, Harper P, Norman A, Joffe JK, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil v fluorouracil, doxorubicin and methotrexate in advanced esophagogastric cancer. J Clin Oncol. 1997;15:261–7.
Acknowledgements
We thank the patients and their families as well as the investigators and site personnel involved in the study.
Author information
Authors and Affiliations
Contributions
RL and SC conceived and designed the study. GR, RL, and SC wrote, edited, and discussed the manuscript. GR, CAC, LC, CC, MP, SM, GL, NS, IB, RC, FZ, DA, AC, SB, DB, and RL gathered the data. CD, ADS, and AC performed statistical analysis. All authors were involved in development, review, and approval of the manuscript. GR, and RL were responsible for the final decision to submit for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical standards
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent was obtained from all patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rosati, G., Cella, C.A., Cavanna, L. et al. A randomized phase III study of fractionated docetaxel, oxaliplatin, capecitabine (low-tox) vs epirubicin, oxaliplatin and capecitabine (eox) in patients with locally advanced unresectable or metastatic gastric cancer: the lega trial. Gastric Cancer 25, 783–793 (2022). https://doi.org/10.1007/s10120-022-01292-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-022-01292-y