
Abstract
Background
The majority of GISTs express mutationally activated KIT. Imatinib and sunitinib are approved KIT-inhibiting therapies. Their efficacy is usually hampered by the acquired multiple secondary drug-resistance KIT mutations. The most problematic resistance subset is GISTs with acquisition of secondary mutations in the KIT activation loop. Here, we establish the spectrum of activity of dasatinib against a comprehensive collection of clinically relevant KIT mutants associated with drug-sensitive and drug-resistant GIST.
Methods
The cellular and in vitro activities of tyrosine kinase inhibitors (TKIs) against mutant KIT were assessed using a panel of engineered and GIST-derived cell lines. The in vivo activities of dasatinib were determined using TKI-resistant xenograft models.
Results
In engineered and GIST-derived cell lines, dasatinib potently inhibited KIT with primary mutations in exon 11 or 9 and a range of secondary imatinib-resistant mutations in exons 13 and 14, encoding the ATP-binding pocket, and in exons 17 and 18, encoding the activation loop, with the exception of a substitution at codon T670. Our data show that dasatinib is more potent than imatinib or sunitinib at inhibiting the activity of drug-resistant KIT mutants. Dasatinib also induces regression in GIST-derived xenograft models containing these secondary mutations. A major determinant of the efficacy of dasatinib for the treatment of advanced GIST is the activity of this inhibitor against KIT mutants.
Conclusion
Dasatinib shows efficacy in cancer models, inhibiting a wide range of oncogenic primary and drug-resistant KIT mutants. These results have implications for the further development of dasatinib precision therapy in GIST patients.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumors (GISTs) are the most common type of sarcoma [1, 2]. Activating mutations of receptor tyrosine kinase KIT are key to the pathogenesis of most GISTs, with nearly 80% of GISTs expressing a mutationally activated KIT receptor [1, 3]. KIT activation not only is essential to the initiation of GISTs but also plays an oncogenic role in tumor maintenance [1, 4, 5]. Accordingly, KIT inhibition has profound effects on GIST viability, given that most GISTs depend upon continuous signaling through mutant constitutively activated KIT oncoprotein [6,7,8,9,10].
Imatinib mesylate, a tyrosine kinase inhibitor (TKI) with potent activity against the pathogenetic KIT kinase, has revolutionized GIST therapy and is currently used as a first-line treatment for metastatic or unresectable GIST [10, 11]. Unfortunately, even those patients with dramatic initial clinical responses ultimately develop resistance to imatinib due to secondary, drug-resistant KIT mutations, resulting in disease progression after 1–3 years of continuous therapy [12,13,14,15,16]. Most cases of secondary resistance are associated with an acquired kinase mutation of the ATP-/drug-binding pocket, encoded by exons 13 and 14, or the kinase activation loop, encoded by exons 17 and 18, on the same allele (cis-conformation) as the primary KIT mutation [1, 13, 14, 17]. The challenge of treating imatinib-refractory GISTs is embarrassed by resistant heterogeneity, as patients can contain polyclonal secondary mutations in distinct tumors, or even within different regions of the same tumor [1, 17,18,19].
Sunitinib malate, another small-molecule TKI with selectivity for KIT and PDGFRA (platelet-derived growth factor receptor A) as well as PDGFRB, VEGFR-1 (vascular endothelial growth factor receptor-1), VEGFR-2, and VEGFR-3, is the standard second-line therapy in GIST patients who were refractory to imatinib or were intolerant of imatinib [7, 14, 17]. Unfortunately, the median PFS (progression-free survival) with sunitinib is approximately half a year [8, 14]. KIT genotyping studies have indicated that sunitinib suppresses KIT ATP-binding pocket mutations (i.e. V654A and T670I, which are resistant to imatinib and sensitive to sunitinib), but has no effect against KIT activation loop mutations (i.e. N822K or Y823D, which are resistant to both imatinib and sunitinib) [14, 17]. Similarly, regorafenib, another multitargeted KIT kinase inhibitor, is a US Food and Drug Administration (FDA)-approved third-line treatment for GIST. Regorafenib treatment offers modest clinical benefit in imatinib-/sunitinib-refractory patients have progressed on imatinib and sunitinib with a median PFS less than half a year [8]. A few imatinib-resistant mutations also lead to sunitinib and regorafenib cross-resistant, thereby explaining the short PFS with second-line sunitinib and third-line regorafenib [1, 12, 17]. Currently, the presence of these polyclonal mutations confines the overall survival (OS) of advanced GIST patients.
Dasatinib is a dual SRC/BCR-ABL kinase inhibitor that inhibits other kinase activities, including KIT [20,21,22,23]. Dasatinib is currently in clinical use for the treatment of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia [21, 24]. Recently, dasatinib has been tested in the third-/fourth-line or even first-line setting in GIST, yielding promising results in phase II studies [25, 26]. Schuetze and colleagues reported a clinical trial of dasatinib for advanced GIST resistant to imatinib, where it showed antitumor activity in a subset of imatinib-refractory GIST patients. However, the correlation of GIST genotype and dasatinib response/resistance phenotype in the clinical study was unclear [25].
To dissect the molecular mechanisms responsible for dasatinib activity against TKI-sensitive and TKI-resistant GIST, we explored the agent against a comprehensive collection of clinically relevant KIT primary and resistance mutants expressed by transiently transfected cells. For comparison, we also investigated the efficacy of imatinib and sunitinib in the same experiments. To further verify our findings, we performed head-to-head comparisons of imatinib, sunitinib, and dasatinib in various cell models. Finally, we employed TKI-sensitive and TKI-resistant GIST cell lines and GIST xenograft models to further corroborate our results.
Materials and methods
Reagents and antibodies
For in vitro cell culture studies, dasatinib, imatinib, and sunitinib were dissolved with dimethyl sulfoxide (DMSO), DMSO and phosphate-buffered saline (PBS), respectively. For in vivo animal studies, dasatinib was dissolved in 80 mM citrate buffer (pH 3.1). Imatinib and sunitinib were dissolved in saline. Primary antibodies were p-KITY721 (Cell Signaling Technology (CST), #3391), KIT (Dako, A4502), p-AKTSer473 (CST, #9271), AKT (CST, #9272), p-MAPKThr202/Tyr204 (CST, #9101), MAPK (CST, #9102) and GAPDH (Sigma, G8795).
Mouse GIST xenografts
Female BALB/c nude mice (5–6 weeks old; weight 18–25 g) were obtained from Shanghai Lingchang BioTech Co. Ltd, PR.China. All animals were maintained in the specific pathogen-free (SPF) facility of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences according to the international, national and institutional guidelines for humane animal treatment and complied with relevant legislation. The mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institutes for Biological Sciences (Approval number SIBS-2017-WYX-1). All mice were acclimatized for at least 7 days before initiation of study. The laboratory mice had free access to food, water and enrichment. The temperature, humidity, light cycle, and cage size were according to laboratory mouse guidelines and regulations. The mice were housed 3–5 mice per cage. Prior to implantation, GIST cells were harvested during exponential growth. Cells (3 × 106) in PBS were formulated as a 1:1 mixture with Matrigel (BD Biosciences) and were subcutaneously injected into 5–6-week-old female BALB/c nude mice. The resulting tumors were measured once per 4 days. Tumor volume was calculated using the formula: tumor volume (mm3) = [(W2 × L)/2] in which width (W) is defined as the smaller of the two measurements and length (L) is defined as the larger of the two measurements. When the average tumor volume reached approximately 75 mm3, the mice were randomized into treatment groups with five mice per group. Mice were treated twice daily by oral gavage with compound or vehicle for 16 days. For animal studies, dasatinib was formulated in 80 mM citrate buffer (pH 3.1). Imatinib and sunitinib were dissolved in saline. Tumor volume was used to evaluate antitumor activity.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA). The difference between two groups was analyzed by paired-sample t test. Data are presented as mean ± standard deviation (SD). Differences between groups were assessed by a paired t test and accepted as significant at p < 0.05. Significance is indicated by asterisk. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Comparative biochemical activity of dasatinib versus imatinib or sunitinib for inhibiting KIT activity in HEK293T transient transfection cells
To evaluate the spectrum of activity of dasatinib against KIT mutants found in GIST patients, we profiled dasatinib biochemically against a comprehensive panel of KIT primary and acquired resistance mutants. We initially tested the efficacy of dasatinib against the most common KIT mutants identified in GIST, namely, KIT exon 11 mutants and KIT exon 9 mutants, using an isogenic, HEK293T transient transfection model. We started with the representative KIT exon 11 V559D as a primary mutation, because this missense mutation is one of the most frequent juxtamembrane domain aberrations identified in GIST. Similarly, the KIT exon 9 A502_Y503dup mutation is the most common extracellular domain (exon 9) mutation found in GIST. Imatinib and sunitinib were used as the comparator TKIs.
Dasatinib suppressed the KIT exon 11 V559D kinase, with an IC50 of < 100 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\) (Fig. 1a, Supplementary Table 1), making it comparable to imatinib (IC50 = 110 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\)) and sunitinib (IC50 = 480 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\)). The biochemical activity of dasatinib for inhibiting the KIT exon 9 A502_Y503dup kinase was more potent than imatinib, with an IC50 of less than 100 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\), whereas the IC50 for imatinib was 250 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\) (Fig. 1i, Supplementary Table 1). Sunitinib had similar potency against this mutant (IC50 < 100 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\)) (Fig. 1i, Supplementary Table 1).

Comparative biochemical activity of dasatinib versus imatinib/sunitinib for inhibiting KIT activity in transiently transfected cells. HEK293 cells were transiently transfected, and protein lysates from transfected cells were prepared and subjected to western blotting using phospho-KIT or total KIT antibody. Representative results for the comparative activity of dasatinib, imatinib, or sunitinib for inhibiting an isolated KIT exon 11 mutant (V559D) (a), an isolated KIT exon 9 mutant (A502_Y503dup) (i), or compound mutations of V559D (b–h) or A502_Y503dup (j) with a secondary imatinib-resistance mutation (V654A, T670I, D816H, D820A, N822K, Y823D, or A829P) are shown. The heterogeneous secondary resistance mutations in exons 17 and 18, encoding the activation loop, are uniformly sensitive to dasatinib in vitro
The effect of secondary drug-resistant mutations on each TKI was next evaluated in the KIT exon 11 V559D backbone. All the reported KIT amino acid residues at which substitutions cause imatinib resistance were evaluated. Imatinib potency was dramatically decreased in the presence of secondary ATP-binding pocket (i.e. V654A encoded by exon 13 and T670I encoded by exon 14) and activation loop (A-loop) mutants (i.e. D816H, D820A, N822K, Y823D and A829P encoded by exons 17 and 18), with IC50 values ranging from 500 to more than 5000 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\) (Fig. 1b–h, Supplementary Table 1). Sunitinib effectively overcame the ATP-binding pocket, but not the A-loop mutants, as expected. The IC50 for sunitinib against exon 11 mutant with secondary V654A, T670I, D816H, D820A, N822K, Y823D, or A829P mutations was 480, 320, > 5000, > 5000, > 5000, > 5000, and > 5000 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\), respectively (Fig. 1b–h, Supplementary Table 1). On the contrary, secondary mutations within the A-loop and the V654A mutation were sensitive to dasatinib. The IC50 for dasatinib against exon 11 mutant with secondary D816H, D820A, N822K, Y823D, A829P, or V654A mutations was 100, < 100, 110, 200, < 100 and 450 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\), respectively (Fig. 1b, d–h, Supplementary Table 1). However, dasatinib had no activity against the gatekeeper (T670I) mutant (IC50 values > 5000 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\)) (Fig. 1c, Supplementary Table 1).
A-loop mutants are most problematic in GIST targeted therapy. To extend the above assessment, the effects of the N822K secondary mutation were further profiled in the exon 9 A502_Y503dup primary mutation backbone. Although some KIT exon 9-mutant GISTs show primary resistance to imatinib, in a few patients, secondary imatinib resistance develops through the acquisition of KIT mutations that are identical to those seen in imatinib-resistant tumors with KIT primary exon 11 mutations. We explored the potency of dasatinib against KIT exon 9-mutant kinases with a secondary N822K mutation and found IC50 value of 200 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\). The IC50 values for imatinib and sunitinib against exon 9-mutant kinases with secondary N822K mutations were > 5000 \(\mathrm{n}\mathrm{m}\mathrm{o}\mathrm{l}/\mathrm{L}\) for both (Fig. 1j, Supplementary Table 1). The relative comparison of the efficacy of the three TKIs (dasatinib versus imatinib and sunitinib) against various KIT mutants, including primary alone as well as primary with secondary mutants, is shown in Supplementary Table 1.
Potency of dasatinib versus imatinib or sunitinib for inhibition of the KIT oncoprotein and KIT signaling pathways in a GIST cell context
To further confirm the above findings in a GIST cell context, we utilized GIST cell lines to assess the biochemical activity of dasatinib for inhibiting the KIT oncoprotein and KIT signaling pathways. Dasatinib was investigated against a panel of GIST cell lines with various KIT primary and secondary mutations (Supplementary Table 2). These included three imatinib-sensitive cell lines (GIST430, GIST-T1 and GIST882), one imatinib-resistant cell line (GIST430/654, with secondary V654A mutation), and one multiple TKI-resistant cell line (GIST48, with secondary D820A mutation).
The ability of dasatinib to inhibit KIT signaling was assessed in the three imatinib-sensitive cell lines (GIST430, GIST-T1, and GIST882). In agreement with our data against the exon 11 V559D mutant expressed in HEK293T cells, dasatinib had equal potency against the KIT exon 11 mutant GIST-T1 and GIST430 cells and exon 13-mutant GIST882 cells, with phospho-KIT inhibition IC50 values of 5, 25 and 25 nmol/L, respectively (Table 1, Fig. 2a–c). The imatinib IC50 for these cell lines was 50, 50 and 100 nmol/L, respectively, which indicated that the potency of dasatinib was better than that of imatinib. In addition, dasatinib was potent at inhibiting activation of the KIT-dependent signaling pathway, as tested by downregulation of downstream signaling molecules phospho-MAPK (T202/Y204) and phospho-AKT (S473) (Fig. 2a–c).

Comparative activity of dasatinib versus imatinib/sunitinib for inhibition of the KIT kinase and activation of downstream signaling pathways in GIST cell lines. GIST cell lines were treated with dasatinib or imatinib/sunitinib for 4 h and then harvested for protein lysates. Whole-cell lysates were immunoblotted, and the membrane was probed with antibodies to activated (p-KIT, p-AKT, and p-MAPK) and total forms of KIT, AKT, and MAPK. The bottom frame contains the results for GAPDH, which was used as a loading control
The GIST430/654 cell line, established from a GIST patient refractory to imatinib, harbors the primary KIT exon 11 mutation (V560_L576del) and the secondary exon 13 V654A missense mutation [27]. The IC50 for imatinib was > 500 nmol/L. As anticipated, this concentration was 0-fold higher than that necessary to suppress exon 11 mutant oncoprotein in the GIST-T1 cells. On the contrary, dasatinib was a significantly more potent agent of the KIT mutant in GIST430/654 cells, with an IC50 of 200 nmol/L. Consistent with the above data, dasatinib shows more effective than imatinib for the suppression of phospho-MAPK and phospho-AKT (Fig. 2d).
The GIST48 cell line, also established from a GIST patient refractory to imatinib, contains the primary KIT exon 11 mutation (V560D) mutation and the secondary exon 17 D820A missense mutation [27]. The imatinib IC50 for GIST48 was > 500 nmol/L. In contrast, dasatinib was substantially more effective than imatinib, with an IC50 of 100 nmol/L. Likewise, dasatinib treatment was significantly more potent than imatinib at suppressing phosphorylation of MAPK and AKT (Fig. 2e).
Dasatinib inhibits proliferation in KIT-driven GIST cell lines
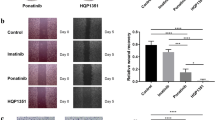
Dasatinib was explored against a panel of GIST cell lines with various primary KIT mutations and imatinib-resistance mutations (Supplementary Table 2). Dasatinib was demonstrated to decrease proliferation of all imatinib-sensitive (GIST430, GIST-T1, and GIST882), imatinib-resistant (GIST430/654), and multiple TKI-resistant (GIST48) cell lines with superior potencies (Fig. 3a–e, Supplementary Table 2), which correlate well with the IC50 values determined by biochemical inhibition of the KIT kinase for these cell lines. No suppression was evidenced observed in the KIT-negative/independent GIST cell line (GIST48B), confirming that the major determinant of the efficacy of dasatinib for the treatment of GIST is the inhibition of this drug against oncogenic KIT mutants (Fig. 3f).

Cell Titer-Glo ATP-based proliferation assays in imatinib-sensitive (GIST-T1, GIST430 and GIST882) (a–c) and imatinib-resistant GIST cell lines (GIST430/654 and GIST48) (d, e) after treatment with dasatinib versus imatinib/sunitinib. All cells were assessed after 3 or 6 days of treatment, with the data normalized to DMSO-only controls. Little activity was observed in the KIT-negative/independent GIST cell line (GIST48B) (f), confirming that the major determinant of the efficacy of dasatinib for the treatment of advanced GIST is the activity of this agent against KIT mutants
Dasatinib substantially abrogates the growth of both imatinib-resistant and multiple TKI-refractory GIST xenograft models
Using a GIST xenograft from GIST48 cells harboring KIT exons 11/17 (V560D/D820A) genomic aberrations, we assessed the antitumor effects of dasatinib. As expected, this xenograft model was refractory to imatinib (Fig. 4a). The results correlates well with the clinical observations, because patients with this KIT exons 11/17 aberrations often show resistance to imatinib and sunitinib therapies. In contrast, dasatinib at doses of 30 mg/kg led to GIST regression and was well tolerated (Fig. 4a). Finally, another GIST xenograft model from GIST430/654 cells harboring the exons 11/13 (V560_L576del/V654A) double alterations was investigated. Imatinib showed no effect on this xenograft model as anticipated (Fig. 4b). Dasatinib at the dose of 30 mg/kg caused substantially GIST regression (Fig. 4b). These results confirm the in vivo antitumor activity of dasatinib against KIT ATP-binding pocket and A-loop mutants, revealing broad activity of dasatinib across the clinically associated KIT mutational spectrum identified in patients.

Antitumor activities of dasatinib versus imatinib/sunitinib as single agents against imatinib-resistant and multiple TKI-resistant GIST xenografts. Mice bearing GIST48 (a) and GIST430/654 (b) cells were treated with dasatinib at 30 mg/kg or imatinib at 50 mg/kg twice a day orally. Control tumor-bearing animals received vehicle. Relative tumor volume (RTV) is shown. All data points are mean ± SE of 5 animals per group. Statistical significance, calculated using Student’s t test in which each treatment group was compared with its vehicle control, is indicated by asterisk. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
GISTs are life threatening in unresectable or metastatic patients. In many patients with advanced GISTs resistant to imatinib, second-line sunitinib and third-line regorafenib therapies manifest limited disease control; hence, more meaningful treatments are urgently needed for most patients [1, 5, 12].
Treatment outcomes and principles for patients with GIST and CML share many important features. Both cancers are initiated by “driver” kinase with KIT for GIST and BCR-ABL for CML. Front-line therapy with the KIT/BCR-ABL inhibitor imatinib causes high response rates. Acquired resistance to TKI in both cancers most commonly involves point mutations in the original kinase target [27]. In CML, several potent BCR-ABL inhibitors, such as nilotinib and dasatinib, have been developed that can be effective therapies for patients’ refractory to imatinib [23, 27,28,29]. These inhibitors also cause higher response rates than imatinib even in the first-line setting [29, 30] and induce a narrower spectrum of secondary drug-resistance mutants [31]. Strikingly, these properties were predicted by preclinical investigations that demonstrated the superior potency of these drugs over imatinib and their ability to suppress many KIT mutants [32].
Given the potential toxicities and the highly variable response to TKIs, as well as the significant economic cost of these agents, there is an urgent need for genomic information that can predict TKI response. Limited studies of the potency of dasatinib against KIT mutants associated with drug resistance has been performed so far [33, 34]. Specifically, dasatinib has only been assessed against the D816 KIT secondary mutations [33]. These D816 mutations are quite rare in GIST but are identified in > 90% of patients with systemic mastocytosis [33, 35]. GIST patients with diseases associated with KIT activation loop mutant kinase have had few meaningful treatment options. Dasatinib exhibits uniform activity in all the KIT activation loop mutants tested in our studies, suggesting that dasatinib may be able to prevent the emergence of the imatinib-/sunitinib-resistant activation loop mutants.
Dasatinib has a broader spectrum of activity against drug-resistant KIT mutations than imatinib or sunitinib, thus accounting for the superior potency of dasatinib in the treatment of patients’ refractory to imatinib or sunitinib. Employing an isogenic model to express isolated or compound KIT mutations, we assessed the IC50 activity of dasatinib across a large panel of clinically associated mutants. Dasatinib is a potent inhibitor of KIT exon 11-/9-mutant kinases with a potency that was equal to or greater than that of imatinib/sunitinib (see Fig. 5a, Supplementary Table 1 for comparative IC50 activity). Dasatinib is substantially more potent than imatinib against the KIT kinase with a secondary mutation of the ATP-binding pocket (V654A) and would be expected to be clinically effective against this mutation. In contrast, this dasatinib-sensitive V654A mutant is resistant to regorafenib (Fig. 5b). Dasatinib is also potent against secondary KIT mutations involving the activation loop. Among the three inhibitors that we tested, dasatinib had the broadest spectrum of activity against drug-resistant mutants (Supplementary Table 1 for comparative IC50 activity, Fig. 5b).

Summary of dasatinib potency against primary oncogenic (a) and imatinib-resistant (b) KIT mutants in GIST. Green light denotes sensitive; red light denotes resistant; orange light denotes intermediate. Potency profile of dasatinib versus imatinib/sunitinib was based on this study, whereas the profile of regorafenib was retrieved from prior reports [38,39,40,41]
In imatinib-refractory GISTs, the frequency of ATP-binding pocket and activation loop mutations is almost equal [1, 17]. What is more, any given patients usually have multiple TKI-resistant lesions, and these tumors can harbor different resistant mutations [17,18,19]. Hence, sunitinib may only inhibit approximately half of the tumors in any individual patient (i.e., the lesions with secondary mutations in the ATP-binding pocket). Based on our data, dasatinib was predicted to have efficacy against the majority of imatinib-resistant GISTs, with the exception of those tumors with a secondary mutation of KIT codon 670, which accounts for approximately 10% of imatinib refractory GISTs, showing sensitive to regorafenib (Fig. 5b). Therefore, our studies show that dasatinib is a more potent TKI for second-line therapy of imatinib-refractory GISTs than the current standard drug, sunitinib. This prediction is also evidenced by the recently reported clinical activity of dasatinib in the third-/fourth-line setting, which is comparable with that seen with the standard second-line sunitinib setting.
Dasatinib is a multitargeted kinase inhibitor of KIT, PDGFRA, SRC, BCR-ABL. The role of SRC has been most extensively characterized in colorectal cancer, and many experiments have demonstrated that increased SRC activity is associated with tumor progression, advanced stage of disease, poor prognosis and poor overall survival [22, 36, 37]. Rotert et al. reported that SRC activity is increased in GIST compared with normal tissue on a small number of patients (29 patients) study. In contrast to most other tumor entities, SRC activity in GIST does not correlate with poor clinical outcome, but decreases during the progression from primary tumor to recurrence and metastasis [38]. SRC activity is associated with longer overall survival in GIST [38]. These observations support the hypothesis that elevated SRC activity seems not an oncogenic driver to promote GIST progression. To completely understand the role of SRC in GIST and the molecular mechanisms behind their clinical behavior, further studies are warranted.
In conclusion, we build the spectrum of activity of dasatinib against different KIT mutants associated with drug-sensitive and drug-resistant GIST. Dasatinib exhibits uniform activity in the KIT activation loop mutants, while there is no activity against the KIT T670I gatekeeper mutant. These results have implications for the further development of dasatinib precision therapy in GIST patients.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information file. Further details are available from the corresponding author on reasonable request.
References
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78.
Niinuma T, Suzuki H, Sugai T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl Gastroenterol Hepatol. 2018;3:2.
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80.
Rossi S, Gasparotto D, Toffolatti L, Pastrello C, Gallina G, Marzotto A, et al. Molecular and clinicopathologic characterization of gastrointestinal stromal tumors (GISTs) of small size. Am J Surg Pathol. 2010;34:1480–91.
Nishida T, Doi T, Naito Y. Tyrosine kinase inhibitors in the treatment of unresectable or metastatic gastrointestinal stromal tumors. Expert Opin Pharmacother. 2014;15:1979–89.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38.
Demetri GD, Reichardt P, Kang Y-K, Blay J-Y, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302.
Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247–58.
Demetri GD, von Mehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, et al. NCCN Task Force Report: Update on the Management of Patients with Gastrointestinal Stromal Tumors. J Natl Compr Canc Netw. 2010;8:S1–41.
von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Casper ES, et al. Gastrointestinal Stromal Tumors, Version 2.2014. J Natl Compr Canc Netw. 2014;12:853–62.
von Mehren M, Joensuu H. Gastrointestinal Stromal Tumors. J Clin Oncol. 2018;36:136–43.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J Clin Oncol. 2008;26:5352–9.
Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64:5913–9.
Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–9.
Gramza AW, Corless CL, Heinrich MC. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin Cancer Res. 2009;15:7510–8.
Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74.
Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R, et al. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005;6:249–51.
Keating GM. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs. 2017;77:85–96.
Brattas MK, Reikvam H, Tvedt THA, Bruserud O. Dasatinib as an investigational drug for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults. Expert Opin Investig Drugs. 2019;28:411–20.
Scuoppo C, Wang J, Persaud M, Mittan SK, Basso K, Pasqualucci L, et al. Repurposing dasatinib for diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2019;116:16981–6.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401.
Efficace F, Stagno F, Iurlo A, Breccia M, Cottone F, Bonifacio M, et al. Health-related quality of life of newly diagnosed chronic myeloid leukemia patients treated with first-line dasatinib versus imatinib therapy. Leukemia. 2019;. https://doi.org/10.1038/s41375-019-0563-0.
Schuetze SM, Bolejack V, Thomas DG, von Mehren M, Patel S, Samuels B, et al. Association of dasatinib with progression-free survival among patients with advanced gastrointestinal stromal tumors resistant to imatinib. JAMA Oncol. 2018;4:814–20.
Montemurro M, Cioffi A, Domont J, Rutkowski P, Roth AD, von Moos R, et al. Long-term outcome of dasatinib first-line treatment in gastrointestinal stromal tumor: a multicenter, 2-stage phase 2 trial (Swiss Group for Clinical Cancer Research 56/07). Cancer. 2018;124:1449–544.
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66:9153–61.
DiNitto JP, Wu JC. Molecular mechanisms of drug resistance in tyrosine kinases cAbl and cKit. Crit Rev Biochem Mol Biol. 2011;46:295–309.
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–70.
Kantarjian HM, Hochhaus A, Saglio G, De Souza C, Flinn IW, Stenke L, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–51.
Davies J. First-line therapy for CML: nilotinib comes of age. Lancet Oncol. 2011;12:826–7.
Bradeen HA, Eide CA, O'Hare T, Johnson KJ, Willis SG, Lee FY, et al. Comparison of imatinib, mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108:2332–8.
Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT Isoforms associated with human malignancies. Cancer Res. 2006;66:473–81.
Dewaele B, Wasag B, Cools J, Sciot R, Prenen H, Vandenberghe P, et al. Activity of dasatinib, a dual SRC/ABL kinase inhibitor, and IPI-504, a heat shock protein 90 inhibitor, against gastrointestinal stromal tumor-associated PDGFRA (D842V) mutation. Clin Cancer Res. 2008;14:5749–58.
Kristensen T, Vestergaard H, Moller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J Mol Diagn. 2011;13:180–8.
Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci USA. 1990;87:558–62.
Rotert JV, Leupold J, Hohenberger P, Nowak K, Allgayer H. Src activity is increased in gastrointestinal stromal tumors-analysis of associations with clinical and other molecular tumor characteristics. J Surg Oncol. 2014;109:597–605.
Serrano C, Marino-Enriquez A, Tao DL, Ketzer J, Eilers G, Zhu M, et al. Correction: complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;121:281.
Smith BD, Kaufman MD, Lu WP, Gupta A, Leary CB, Wise SC, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35:738–51.
Garner AP, Gozgit JM, Anjum R, Vodala S, Schrock A, Zhou T, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20:5745–55.
George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30:2401–7.
Acknowledgements
The authors thank the professor Jonathan A. Fletcher at Harvard Medical School for GIST882, GIST48, GIST48B, GIST430 and GIST430/654 cell lines.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81572642 to YW), the National Key R & D Program of China (2016YFC1302100), and the Basic Research Project of Shanghai Science and Technology Commission (16JC1405600 to YW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
L Duan is an employee of Chia Tai Tianqing Pharmaceutical Group Co. Ltd. No competing interests were disclosed by the other authors.
Ethical approval
All animals were maintained in the specific pathogen-free (SPF) facility of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences according to the international, national and institutional guidelines for humane animal treatment and complied with relevant legislation. The mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institutes for Biological Sciences (Approval number SIBS-2017-WYX-1).
Informed consent
All authors give consent for the publication of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zeng, C., Zhu, L., Jia, X. et al. Spectrum of activity of dasatinib against mutant KIT kinases associated with drug-sensitive and drug-resistant gastrointestinal stromal tumors. Gastric Cancer 23, 837–847 (2020). https://doi.org/10.1007/s10120-020-01069-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-020-01069-1