
Abstract
The purpose of this study was evaluation of the VAPChip assay based on the “Rapid-Array-PCR-technology” which targets 13 respiratory pathogens and 24 β-lactam resistance genes directly on respiratory clinical specimens. The first step included analysis of 45 respiratory specimens in order to calibrate and determine the threshold for target genes. The second prospective step involved 85 respiratory samples from patients suspected of nosocomial pneumonia collected in two academic hospitals over an 8-month period. Results of the VAPChip assay were compared to routine methods. The first step showed a large proportion of positive signals for H. influenzae and/or S. pneumoniae. For identification, discrepancies were observed in seven samples. Thresholds were adapted and two probes were re-designed to create a new version of the cartridge. In the second phase, sensitivity and specificity of the VAPchip for bacterial identification were 72.9% and 99.1%, respectively. Seventy (82%) pathogens were correctly identified by both methods. Nine pathogens detected by the VAPChip were culture negative and 26 pathogens identified by culture were VAPChip negative. For resistance mechanisms, 11 probes were positive without identification of pathogens with an antimicrobial-susceptibility testing compatible by culture. However, the patient’s recent microbiological history was able to explain most of these positive signals. The VAPChip assay simultaneously detects different pathogens and resistance mechanisms directly from clinical samples. This system seems very promising but the extraction process needs to be automated for routine implementation. This kind of rapid point-of-care automated platform permitting a syndromic approach will be the future challenge in the management of infectious diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nosocomial pneumonia (NP), including hospital-acquired pneumonia (HAP) and ventilator-associated pneumonia (VAP), is one of the most prevalent health-care associated infections causing high morbidity and mortality [1]. Indeed, VAP affects 10–25% of intubated patients in intensive care units (ICUs) and it is associated with high mortality rates reaching more than 50% [1,2,3].
Several studies have established that rapid and adequate empirical treatment improves patient outcome in patients affected by NP [3,4,5]. The aim for critical care physicians managing patients with a suspected pneumonia is to provide an appropriate treatment (i.e. adapted to the aetiological agent(s) and its/their antibiotic susceptibility profile) as early as possible in the course of the infection [6]. Because of delayed conventional bacteriologic diagnosis (48–72 h turn-around time), current standards of treatment for NP rely on empirical, probabilistic selection of antibiotics. This approach often leads to overuse of costly broad spectrum antibiotics, which contribute to excess cost of care, toxicities and the further selection of multiresistant bacteria.
The causative pathogens of severe infections have a clear impact on outcome, with higher mortality rates caused by multidrug-resistant organisms (MDRO) [2, 3]. The spread of these MDRO in the hospital setting is considered a global threat, particularly for the ICU patients who are predominantly exposed to these very harmful bacteria [7]. The effective treatment of infections is becoming more and more challenging due to the increasing number of infections caused by these multi-resistant pathogens. In this context, accurate and timely identification of pathogens with their antimicrobial susceptibility pattern could significantly reduce the time to targeted therapy, thus reducing antimicrobial exposure, hospital length of stay, and health costs.
Currently, the available microbiological tests rely on time-consuming methods such as culture, followed by bacterial identification and antimicrobial susceptibility testing (AST). The VAPChip assay (Eppendorf technology®) based on the “Rapid Array-PCR technology” (Rap-ID) combines the sensitivity, speed (4 h) and quantification capacities of real-time PCR (qPCR) with the ability to detect simultaneously a wide range of target genes by using the DNA array methodology (multiplexing) [8]. In the VAPChip assay, PCR and detection of amplification products through DNA-array hybridisation are performed simultaneously in the same analytical closed vessel. The assay targets 13 bacterial species and 24 resistance genes associated with beta-lactam resistance encoding carbapenemases, extended-spectrum β-lactamases and penicillin binding protein 2a (PBP2a) [8]. In the preliminary study performed on a large collection of reference isolates, the authors showed that the limit of detection of the assay was between 10 and 100 genome copies/PCR with a sensitivity and specificity ranging from 95.8% to 100% for species identification and detection of resistance genes [8].
The aim of this proof-of-concept study was to evaluate the diagnostic performance of the VAPChip assay for the detection and characterization (species identification and detection of resistance mechanisms) of the etiologic agents causing NP directly on respiratory samples in patients admitted to the ICU.
Materials and methods
Study design
The study was conducted in two steps: (1) calibration and determination of the threshold for normalized identifications (Ct-based), including a prospective analysis in the ULB-Erasme hospital of 45 respiratory tract samples, and (2) microbiological validation. This second step was a prospective, observational study conducted at two tertiary care referral hospitals (ULB-Erasme and UCL-Mont-Godinne) over an 8-month period. The second step compared a modified VAPChip assay based on the first phase evaluation, with routine culture of lower respiratory tract samples.
Inclusion criteria of respiratory samples
For the first step, residual aliquots of respiratory specimens from hospitalized adult patients (≥ 18 years) and of a good quality (Gram staining with <25 epithelial cells and >25 white blood cells by field or <25 epithelial cells and <25 white blood cells by field) were selected. Duplicate specimens originating from the same patients were excluded. These respiratory samples included sputum, endotracheal or endobronchial aspirates, and bronchoalveolar lavages (BAL) delivered to the laboratory between 8 am to 3:30 pm from Monday to Friday.
For the second step additional criteria were used; patients hospitalized in the ICU of one of the two academic hospitals for more than 48 h and suspected of pneumonia were prospectively included. Pneumonia was defined by the presence of either a new or progressive infiltrate on chest radiography and at least two of the following clinical features: temperature > 38 °C or <36 °C; hyperleucocytosis >10,000 cells/ml or leucopenia <5000 cells/ml; purulent respiratory secretions; and gas exchange degradation (hypoxemia) [9]. Other information, including the date of hospital and ICU admission, patient medical history, current or recent antimicrobial therapy (within 7 days) and microbiological culture results of previous (within 7 days) or current samples, was collected.
Microbiological methods
All respiratory samples, except BALs, were homogenized and diluted at 1:1 in dithithreitol (sputasol®). All samples were split into three parts: one for the routine diagnostic tests, one for the VAPChip assay and one was stored at 4 °C for 1 week for further analysis if needed (in case of discrepancies).
Routine diagnostic tests
Respiratory samples were cultured quantitatively onto selective and non-selective media (blood, chocolate, CAP and MacConkey agar plates, BioMerieux) and cultures were examined daily for 48 h. Presumed pathogens were identified by MALDI-TOF mass spectrometry (Brucker Daltonics) and/or by additional phenotypic tests (e.g. optochin, Vitek 2). AST was determined by disk diffusion methods according to CLSI recommendations [10].
VAPChip assays
DNA extraction step
For the DNA extraction, 500 μl of sputolized (sputum, AET) or non sputolized samples (BAL) were mixed with 1 ml of NAC buffer (10 ml citrate du sodium 2.94%, 10 ml NaOH 4%, 100 mg N-acetyl-L-cysteine). The lysis step was performed by using the LightCycler® Advanced Lysis Kit and the MagNa Lyser insturment (Roche Diagnostics®, Penzberg, Germany) according to the manufacturer’s instructions. The DNA purification step was performed by using the DNeasy Blood Tissue Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s instructions.
The VAPChip assays were performed and interpreted in both centers following manufacturer’s instructions as previously described (Eppendorf Array technologies; www.eppendorf.com). [8]
Definitions, interpretation of results and discrepancies analysis
A negative VAPChip result was defined as the absence of any significant signal for identification (ID) or for any of the targeted resistance genes. A positive VAPChip was defined as a positive signal for ID and/or any of the targeted resistance genes.
A culture result was considered as negative or non significant when no growth was observed or in the presence of polymicrobial oral flora (composed of orophayngeal commensal species). For bacterial species targeted by VAPchip, culture results were considered as positive when: (i) the amount of colonies was superior to the cut-off defined as ≥106 CFU/ml for sputum and aspirates and ≥104 CFU/ml for BALs [11, 12], (ii) growth of any amount of colonies for L. pneumophila, and (iii) growth of amount of colonies inferior to the cut-off of significance but visualized at microscopic direct examination or showing a monomorphic aspect of the culture.
Susceptible bacterial isolates were defined as no β-lactam resistance phenotype detected by AST, and resistant bacterial isolates were defined as any β-lactam resistance mechanism detected by AST. The antibiogram phenotype was confirmed by molecular analysis only in case of discrepancy between VAPChip and AST.
Results were interpreted as either concordant or discrepant.
For identification, results were considered concordant when ID signal obtained by VAPChip matched with the pathogen detected by culture. Discrepant results were defined as a positive ID signal by VAPChip showing a negative culture or vice versa. The sensitivity and specificity of identification were calculated for the 13 cultivable bacterial species detected by the VAPChip assay.
For resistance mechanisms to beta-lactam agents, results were considered concordant when in a sample, the VAPChip signal was compatible with AST (limited to resistance mechanisms targeted by the VAPChip) or identical to the gene identified by multiplex PCR on isolates grown by culture. A result was considered discordant when a positive signal was detected by VAPChip but not confirmed by multiplex PCR on culture grown pathogens or when culture was negative.
Discrepant results were solved at three levels: (1) Bacterial level: pure colonies after culture were reanalyzed by VAP-Chip and MALDI-TOF. The presence of resistance genes was confirmed by using several multiplex PCR considered as reference method for genes encoding bla OXA48, bla VIM, bla IMP, bla KPC, and bla NDM or bla TEM, bla SHV, bla CTX or bla OXA23, bla OXA24 and bla OXA58 for gram negative rods and mecA gene for staphylococci. (2) Sample level: clinical respiratory samples were analyzed by a home-brew real-time PCR (qPCR) to confirm VAPChip results. The qPCR allowed confirmation of bacterial identification and of resistance genes to beta-lactams antibiotics. (3) Patient level: results of microbiological analysis of other clinical specimens from the same patient within the previous 7 days were reviewed seeking colonization or infection by a respiratory pathogen showing an AST phenotype compatible with the VAPChip signal.
Results
First step
During the first step of the study (calibration phase), a total of 45 samples (24 sputum and 21 endobronchial aspirates) were included. Results of the bacterial culture and the VAPChip before and after calibration are shown in Table 1. The VAPchip analysis showed a large proportion of positive signals for H. influenzae (n = 12) and/or S. pneumoniae (n = 12) in 25 samples with non significant culture results for these two pathogens. Therefore, the threshold values for H. influenzae and S. pneumoniae were adapted in order to improve the discrimination between oropharyngeal contamination and significant bacterial load responsible for infection. The positive threshold was adapted at <29 and <26 detection cycles (Ct) for H. influenzae and S. pneumoniae, respectively. Those threshold values correspond to about ≥104 CFU/ml. After the threshold modification, 22 of 24 signals became negative in these samples for these two pathogens.
By culture, 17 samples were negative and 28 samples yielded growth of 37 bacterial isolates targeted by the VAPChip.
For VAPChip, 41 bacterial signals tested positive in 30 samples, and the 15 remaining samples were negative (Table 1).
For bacterial identification, seven samples showed discrepancies (Table 1). The VAPChip assay missed one S. aureus which showed after sequencing three SNPS at the 16S sequence corresponding to the hybridization site of the PCR primers used by the VAPChip assay. The VAPChip detected two K. pneumoniae not recovered by culture. Two C. koseri recovered by culture were misidentified by VAPChip as K. oxytoca. Concerning the antimicrobial resistance mechanisms, one methicillin-susceptible S. aureus (MSSA) isolate was misinterpreted as methicillin-resistant (MRSA) by the VAPChip. Moreover, in nine samples, the VAPChip showed a positive signal for the mecA gene but not for S. aureus, suggesting the presence of methicillin-resistant coagulase-negative staphylococci (MR-CNS) in those samples. In order to improve the cartridge, the probes for detection of K. oxytoca and S. aureus were re-designed and a specific probe to detect CNS was included. This new version of the cartridge was further evaluated in the second step of the study.
Second step
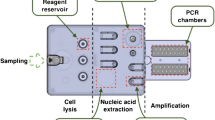
During this second step (validation phase), a total of 85 samples (14 BAL, 62 endobronchial aspirates and 9 sputa) were included (45 from ULB-Erasme site and 40 from UCL-Mont-Godinne site) (Fig. 1). Eighteen samples were both VAPChip and culture-negative. Sixty-seven samples were positive by culture and/or by VAPChip (Table 2). The culture-positive samples (n = 64) yielded growth of 96 significant pathogens included in the VAPChip panel. Seventy out of the 96 culture positive pathogens were properly identified from 55 samples by both methods, while 26 showed discrepant results. These 26 pathogens (from 25 samples) detected by culture were VAPChip negative (Fig. 1 and Table 2). However, four of the six H. influenzae recovered by culture were weakly detected by VAPChip (Ct slightly above the threshold). The VAPChip was repeated on these 26 bacterial isolates and all isolates were well assigned by VAPChip. Moreover, nine pathogens detected by the VAPChip from seven samples were not found in culture. Three of these seven patients were known to be colonized/infected by the pathogens detected by VAPChip and five were under antimicrobial therapy at the time of sampling, thus partially explaining the negative culture. For bacterial identification, the sensitivity and specificity of the VAPChip was 72.9% and 99.1%, respectively.

Screening of respiratory tract samples
Concerning the antimicrobial resistance mechanisms, 17 signals were positive by VAPChip for a resistance gene. For gram-positive cocci (GPC), eight samples showed a positive signal for mecA gene by VAPChip. Five of these were S. aureus culture positive, and a signal for S. aureus by VAPChip was observed. Only three isolates were confirmed as MRSA. In the last two samples, MSSA isolates were recovered by culture, suggesting MRSA false interpretation due to an interference with MR-CNS harboring the mecA gene. For gram-negative rods (GNR) (Table 3), five samples showed positive signals for ESBL genes (CTX-M (n = 3), TEM-ESBL (n = 2)) by VAPChip. By culture, E coli ESBL (CTX-M) were detected in two of these samples, but no Enterobacteriacea ESBL could be demonstrated in the last three samples. One of these three patients had a recent microbiological history of a K. pneumoniae ESBL-producing in respiratory samples. Four samples showed positive signals for carbapenemases genes by VAPChip including VIM (n = 1) and oxa-23 (n = 3). In one sample, A. baumannii OXA-23 were detected by culture. In the three others samples no GNR were detected by culture. However, microbiological history review showed previous positive respiratory samples for VIM-producing P. aeruginosa for one patient and OXA-23-producing A. baumannii for another patient.
Discussion
The VAPChip system is an automated platform based on a RT-PCR array for rapid identification of pathogens and resistance mechanisms [8]. The aim of this proof-of-concept study was to demonstrate the feasibility of a syndromic approach for the diagnosis of nosocomial pneumonia. The prototype showed many advantages including: rapid turn-around time (3-4 h), analysis performed directly on the respiratory samples, and acquisition of semi-quantitative results. In a first study published by Boagaerts et al., the concept was validated on genotypically-characterized bacteria and clinical isolates in order to assess the accuracy of the probes and the limit of detection [8]. However, the cut-off of this new diagnostic system should be further validated in studies similar to this one.
In pulmonary samples, the distinction between colonization and infective organisms remains challenging. The first step of our study was used to improve the VAPChip cartridge, particularly to determine positive threshold values for S. pneumoniae and H. influenzae. The new version of the cartridge was used in the second step. In this validation step, the device was tested in two clinical microbiology laboratories. This evaluation showed that the VAPChip system has a sensitivity of 72.9% and a specificity of 99.1% for the identification of pathogens.
Few (n = 7) samples were VAPChip-positive and culture-negative but 26 cases were VAPChip-negative and culture-positive. However, these samples showed proper identification results after repeating the VAPChip on colonies from the pure culture. This suggests more a problem of sensitivity of the technique than a problem of design of the probes.
For the detection of resistance mechanisms, some probes (n = 11) were VAPChip-positive without identification of pathogens with an AST compatible by culture. The presence of methicillin-resistant SCN could be the cause of the numerous mecA positive signals (n = 5). In order to prevent MRSA misinterpretation due to the presence of a mixed flora of MSSA and mecA-positive SCN, the VAPChip cartridge should be improved by adding probes detecting, for example, the highly conserved SCCmec chromosomal junction (orfX) and the different SCCmec types. For GNR, some of these positive signals by VAPChip for resistance mechanisms could be explained after reviewing patient’s microbiological histories (n = 3).
Our study presented some limitations. Mainly, a DNA-extraction step was required before launching the VAPChip device. This extraction step was laborious and not automated. In order to use the VAPChip device as a point-of-care system, this step needs to be improved and automated. The lack of an automated DNA extraction system and/or the presence of PCR inhibitors may be responsible for the 26 VAPChip false-negative cases. An improvement in this step seems crucial before implementing this kind of device for routine analysis. The VAPChip threshold values for H. influenzae and S. pneumoniae were defined based on the results of the first step. To be more accurate, this threshold should be reassessed in further studies.
Few authors have already published data about the molecular diagnostics of respiratory pathogens. Jahn et al. and Schulte et al. used the “Prove-it sepsis assay” and the “Curtis Unyvero”, respectively, on 35 and 739 respiratory samples for rapid identification of pulmonary pathogens. The sensitivity of the Prove-it assay and of the Curetis were 87.5% and 70.6%, respectively, while the specificity reached 28.6% and 95.2%, respectively. Both studies did not include the evaluation of the detection of resistance mechanisms [13, 14]. Recently, Gadsby et al. used a multibacterial and multiviral molecular system for the diagnosis of confirmed CAP (community acquired pneumoniae). Their molecular approach improved the detection of pathogens and they postulated that their system has the potential to enable de-escalation from broad-spectrum empirical antimicrobials to pathogen-directed therapy [15]. Jamal et al. tested the Curetis Unyvero system on 49 patients suspected of nosocomial pneumonia. They evaluated the identification of pathogens, the detection of resistance mechanisms and the impact on the management of NP. However, the cultures were not quantitative, patients’ microbiological histories were not reviewed, and even more importantly, the detection of resistance mechanisms was not confirmed by molecular techniques. They concluded that the cumulative agreement for identification of pathogens was 70%. The most common discordance was the detection of pathogens by the Curetis system in culture-negative samples. Nevertheless, the authors concluded that rapid molecular diagnosis was beneficial for patients by allowing fast delivery of appropriate targeted antibiotic therapy [16].
The advantage of the VAPChip is the simultaneous semi-quantitative detection of pathogens and antimicrobial resistance mechanisms. The detection of resistance mechanisms joined with the identification of the organism could be essential for the control of the spread of multidrug resistant bacteria in epidemic contexts. Further investigations are needed to evaluate the impact of this syndromic approach on the quality of the delivered individual medical care, but also on the reduction of antibiotic consumption and overall costs. Moreover, the choice in the panel of genes needs to be adapted to the local epidemiology.
In conclusion, we believe that a syndromic approach using rapid point-of-care systems will be one of the future challenges in the management of infectious diseases. The close collaboration between the clinical microbiologist and the infectious disease physician will be essential for the interpretation of these complicated but rapid molecular results to best adapt patient’s treatments.
References
Rotstein C, Evans G, Born A, Grossman R, Light RB, Magder S, McTaggart B, Weiss K, Zhanel GG (2008) Clinical practice guidelines for hospital-acquired pneumonia and ventilator-associated pneumonia in adults. Can J Infect Dis Med Microbiol 19:19–53
Brusselaers N, Labeau S, Vogelaers D, Blot S (2013) Value of lower respiratory tract surveillance cultures to predict bacterial pathogens in ventilator-associated pneumonia: systematic review and diagnostic test accuracy meta-analysis. Intensive Care Med 39:365–375. https://doi.org/10.1007/s00134-012-2759-x
Endimiani A, Hujer KM, Hujer AM, Kurz S, Jacobs MR, Perlin DS, Bonomo RA (2011) Are we ready for novel detection methods to treat respiratory pathogens in hospital-acquired pneumonia? Clin Infect Dis 52(Suppl 4):S373–S383. https://doi.org/10.1093/cid/cir054
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Townsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb S, Beale RJ, Vincent JL, Moreno R (2013) Surviving sepsis campaign guidelines committee including the pediatric subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 39:165–228. https://doi.org/10.1007/s00134-012-2769-8
Iregui M, Ward S, Sherman G, Fraser VJ, Kollef MH (2002) Clinical importance of delays in the initiation of appropriate antibiotic treatment for ventilator-associated pneumonia. Chest 122:262–268
Burillo A, Bouza E (2014) Use of rapid diagnostic techniques in ICU patients with infections. BMC Infect Dis 14:593. https://doi.org/10.1186/s12879-014-0593-1
Lung M, Codina G (2012) Molecular diagnosis in HAP/VAP. Curr Opin Crit Care 18:487–494. https://doi.org/10.1097/MCC.0b013e3283577d37
Bogaerts P, Hamels S, de Mendonca R, Huang TD, Roisin S, Remacle J, Markine-Goriaynoff N, de Longueville F, Plüster W, Denis O, Glupczynski Y (2013) Analytical validation of a novel high multiplexing real-time PCR array for the identification of key pathogens causative of bacterial ventilator-associated pneumonia and their associated resistance genes. J Antimicrob Chemother 68:340–347. https://doi.org/10.1093/jac/dks392
American Thoracic Society (2005) Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 171:388–416
Clinical and Laboratory Standards Institute (2011) Performance standards for antimicrobial susceptibility testing. CLSI M100-S21, Wayne, PA
Lynne S. Garcia & Associates (2007) Clinical microbiology procedures handbook, volume I, 3rd edn. LSG & Associates, Santa Monica, California, pp 3.11.2.17–20
Cornaglia G (2012) European manual of clinical microbiology (ESCMID, SFM), 1st edition. SFM, Epernay, France, p 158
Jahn K, Kuisma M, Mäki M, Grendelmeier P, Hirsch HH, Tamm M, Papakonstantinou E, Stolz D (2015) Molecular diagnostics for bacterial infections in bronchoalveolar lavage a case-control, pilot study. Swiss Med Wkly 145:w14193. https://doi.org/10.4414/smw.2015.14193
Schulte B, Eickmeyer H, Heininger A, Juretzek S, Karrasch M, Denis O, Roisin S, Pletz MW, Klein M, Barth S, Lüdke GH, Thews A, Torres A, Cillóniz C, Straube E, Autenrieth IB, Keller PM (2014) Detection of pneumonia associated pathogens using a prototype multiplexed pneumonia test in hospitalized patients with severe pneumonia. PLoS One 9:e110566. https://doi.org/10.1371/journal.pone.0110566
Gadsby NJ, McHugh MP, Russell CD, Mark H, Conway Morris A, Laurenson IF, Hill AT, Templeton KE (2015) Development of two real-time multiplex PCR assays for the detection and quantification of eight key bacterial pathogens in lower respiratory tract infections. Clin Microbiol Infect 21:788.e1–788.e13. https://doi.org/10.1016/j.cmi.2015.05.004
Jamal W, Al Roomi E, AbdulAziz LR, Rotimi VO (2014) Evaluation of Curetis Unyvero, a multiplex PCR-based testing system, for rapid detection of bacteria and antibiotic resistance and impact of the assay on management of severe nosocomial pneumonia. J Clin Microbiol 52:2487–2492. https://doi.org/10.1128/JCM.00325-14
Acknowledgements
The authors thank Madjit Taguemont for his technical support.
Funding
This work was supported by a research grant from the Région Wallonne of Belgium (BioWin VAPChip convention 6214).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S.H. and F.D. are employees of Eppendorf Array Technologies. All others: none to declare.
Ethical approval
This clinical study was approved by both of the participating university hospital’s institutional medical ethics committees: ULB-Erasme and UCL-Mont-Godinne (approval number P2011/298). Patient informed consent was not required because the study did not require any additional sample collection, nor changes in therapeutic management.
Additional information
S. Roisin and T-D. Huang contributed equally to this article.
Rights and permissions
About this article

Cite this article
Roisin, S., Huang, TD., de Mendonça, R. et al. Prospective evaluation of a high multiplexing real-time polymerase chain reaction array for the rapid identification and characterization of bacteria causative of nosocomial pneumonia from clinical specimens: a proof-of-concept study. Eur J Clin Microbiol Infect Dis 37, 109–116 (2018). https://doi.org/10.1007/s10096-017-3108-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-017-3108-3