
Abstract
Rapid bacterial species identification and antibiotic susceptibility testing in positive blood cultures have an important impact on the antibiotic treatment for patients. To identify extended-spectrum beta-lactamases (ESBL) directly in positive blood culture bottles, we developed a workflow of saponin extraction followed by a bottom-up proteomics approach using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). The workflow was applied to positive blood cultures with Escherichia coli and Klebsiella pneumoniae collected prospectively in two academic hospitals over a 4-month period. Of 170 positive blood cultures, 22 (12.9%) contained ESBL-positive isolates based on standard susceptibility testing. Proteomic analysis identified CTX-M ESBLs in 95% of these isolates directly in positive blood cultures, whereas no false positives were found in the non-ESBL producing positive blood cultures. The results were confirmed by molecular characterisation of beta-lactamase genes. Based on this proof-of-concept study, we conclude that LC-MS/MS-based protein analysis can directly identify extended-spectrum beta lactamases in E. coli and K. pneumoniae positive blood cultures, and could be further developed for application in routine diagnostics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Infections caused by antibiotic resistant Gram-negative bacteria are an increasing problem worldwide. In the Netherlands, resistance towards third generation cephalosporins through extended-spectrum beta-lactamases (ESBLs) is the most frequently found antibiotic resistance of medical importance [1]. Unrecognised, infections with ESBL-producing bacteria pose a serious threat, as they are associated with high morbidity and mortality rates [2]. Escherichia coli and Klebsiella pneumoniae are reported among the main representatives of ESBL-producing bacteria. In the Netherlands, ESBL prevalence is approximately 10% among infected patients [3].
Extended-spectrum beta-lactamases are a group of beta-lactamases which can also hydrolyze third generation cephalosporins. The detection of these ESBL-enzymes is currently provided indirectly by the results of standard susceptibility testing of cultured bacteria, followed by a phenotypic confirmation assay or a genetic test. Direct detection of the enzyme responsible would provide molecular information regarding the phenotype. Protein analysis by way of mass spectrometry has changed microbiological practice in recent years through the introduction of Matrix-Assisted Laser Desorption Ionisation-Time of Flight Mass Spectrometry (MALDI-TOF MS) for species identification [4]. However, the inherent limitations of these instruments such as the limited dynamic range and resolution limit the general applicability to accurately detect the presence of ESBL, particularly in identifying the nature of the underlying enzyme.
Peptide analysis by bottom-up proteomics is commonly used to directly identify proteins and can be used for in-depth proteomic characterisation of resistant bacteria, often using multi-dimensional protein and/or peptide fractionation techniques [5–7]. However, straight analysis of proteolytic digests of total cellular protein extracts also allows to directly identify resistance-related proteins such as beta-lactamases [8, 9]. This leads to shorter analysis times compared to comprehensive proteome studies while maintaining the inherent specificity of directly identifying the protein of interest. Previously we developed such a proteomic platform for the direct detection of OXA-48 and KPC carbapenemases in bacterial cultures of clinical isolates [10–12].
A significant reduction in analysis time would be achieved when bacterial beta-lactamases could be directly analyzed in positive blood cultures. Therefore, the aim of this study was to develop a LC-MS/MS based bottom-up proteomics workflow to identify ESBL-producing E. coli and K. pneumoniae directly in blood cultures and to test the performance of this workflow in a proof-of-principle study using clinical blood culture samples collected in a prospective study.
Materials and methods
Design of study
This study was designed to evaluate the use of proteomic analysis by LC-MS/MS for the detection of extended-spectrum beta lactamases directly from positive blood culture bottles that grow E. coli or K. pneumonia, during a prospective study. Two academic centers participated in the study: the Erasmus University Medical Center in Rotterdam and the Leiden University Medical Center in Leiden. During a period of 4 months (July–October 2015), all positive blood cultures with K. pneumoniae or E. coli, were included in the study.
Comparison of sample preparation methods
Two methods were evaluated for the analysis of bacterial proteins in blood cultures, serum separator tubes and a differential lysis protocol using saponin. For this purpose, negative blood culture bottles were spiked with different amounts of liquid broth culture of E. coli BL21(DE3) pLysS to mimic different bacterial densities in positive blood cultures.
Serum separator tubes feature a gel through which red blood cells can migrate while bacteria are pelleted on top of the gel. Four mL from a spiked blood culture bottle were applied to the tubes (Becton Dickinson, Breda, The Netherlands) and the mixture was centrifuged at 6000 g for 10 min. Serum was removed and the pellet was washed twice with 1 mL of phosphate buffered saline (PBS) followed by a 5 min centrifugation at 6000 g. The bacterial pellet on top of the gel was resuspended in 100 μL PBS and transferred to an Eppendorf tube. The gel was rinsed with 100 μL PBS another three times to recover any residual bacteria and this was added to the vial. The resulting bacterial suspension was centrifuged at 10,000 g for 1 min and the supernatant was removed. 100 μL of 50% trifluoroethanol (TFE) solution was added for protein extraction and solubilisation. This suspension was sonicated in an ultrasound water bath for 2 min. Suspensions were heated to 60 °C for an hour. The resulting lysates were then subjected to protein digestion.
For the saponin protocol, 4 mL of the spiked blood culture bottle was mixed with 1 mL of a saponin (Sigma-Aldrich, Zwijndrecht, The Netherlands) stock solution (5% w/v, final concentration 1% w/v). The mixture was vortexed, incubated at room temperature for 5 min and centrifuged at 6000 g for 10 min. The cell pellet was washed three times with 1 mL PBS and centrifuged at 10,000 g for 1 min and, following the final centrifugation step, re-suspended in 100 μL 50% TFE solution. The suspension was sonicated using an ultrasound water bath for 2 min. Suspensions were heated at 60 °C for an hour. The resulting lysates were stored at −80 °C until further analysis.
Blood culture and species identification
Blood cultures were drawn as part of normal clinical routine. A sample of 8–10 mL of blood was used per bottle (Bactec Plus Aerobic and Bactec Plus Anaerobic, Becton Dickinson, Breda, The Netherlands) for blood culturing (Bactec FX, Becton Dickinson, Breda, The Netherlands). Bacterial species in positive cultures were identified directly from 1 mL blood culture by MALDI-TOF MS analysis (Microflex, Bruker Daltonics, Bremen, Germany) according to an in-house developed protocol adapted from literature [13]. All positive flagged blood cultures were stored at 4 °C and processed within 48 h using the saponin protocol described above.
Susceptibility testing
Susceptibility testing was performed with VITEK 2 (bioMérieux, Marcy l’Etoile, France). The presence of ESBLs was performed using the combination disk diffusion tests (Rosco Diagnostica A/S, Taastrup, Denmark) according to the Dutch guideline [14]. Disks used were ceftazidime (30 μg), ceftazidime + clavulanate (30 + 10 μg), cefotaxime (30 μg) and cefotaxime + clavulanate (30 + 10 μg). ESBL production was considered positive if the zone diameters around one or both of the combination disks was ≥5 mm compared to the corresponding antibiotic-only disk. E-tests for ceftazidime, cefotaxime and meropenem were performed on Mueller Hinton E agar media according to the recommendations of EUCAST (http://www.eucast.org/clinical_breakpoints/).
In-solution protein digestion
Stored lysates (at −80 °C) were thawed for further processing for bottom-up proteomics. Reduction was performed with dithiothreitol (DTT, final concentration 2.5 mM in 25 mM ammonium bicarbonate) at 60 °C for 15 min. Alkylation was performed in the dark with iodoacetamide (final concentration 5.5 mM in 25 mM ammonium bicarbonate) for 15 min. Following alkylation the samples were digested overnight using sequencing grade modified trypsin (12.5 ng/μl, Promega, Leiden, The Netherlands). The next day the resulting digests were lyophilized and reconstituted in 0.5% trifluoroacetic acid (TFA) for pre-column trapping during LC-MS/MS analysis.
Molecular characterisation
All ESBL positive isolates (n = 22) were analyzed for the presence of beta-lactamase genes. An in-house real-time multiplex bla CTX-M PCR was used for analysis of the specific CTX-M groups. For primer design, an alignment of the available bla SHV gene sequences from GenBank® was made using the AlignX program (Vector NTI Advance 11, Invitrogen). Primers and probes were developed in-house using Beacon Designer (Premier Biosoft, Palo Alto, U.S.A.). Subsequently, beta-lactamase gene bla SHV was amplified using PCR and further investigated by nucleotide sequence analysis [15]. All primers and probes used in this study are listed in Table 1. These molecular assays have been developed and internally validated at the LUMC and are used in daily routine.
LC-MS/MS analysis and data processing
Peptide mixtures were analyzed using nano reversed-phase liquid-chromatography coupled to tandem mass spectrometry (nano LC-MS/MS). The nano-LC system (Ultimate 3000 RSLCnano, Dionex) combines a 2-cm Acclaim PepMap 100 guard column with an Acclaim PepMap RSLC column (C18, 75 μm × 50 cm with 2 μm particles). A multi-step gradient going from 5 to 55% B in 180 min was used (solvent A being 0.1% formic acid in water and solvent B 0.1% formic acid in 80% acetonitrile) at a rate of 300 nl min−1. Mass spectrometry analysis was carried out on a maXis Impact UHR-TOF-MS (Bruker Daltonics) in data dependent MS/MS mode, with precursors ranging from m/z 300–1200. After MS/MS analysis precursors were excluded from selection dynamically for one minute.
Raw data were converted to Mascot Generic Files (MGF) and analyzed by database searching using the Mascot algorithm (Mascot 2.5.1, Matrix Science, London, UK) using Mascot Daemon 2.5.1. To ensure a comprehensive search of all beta-lactamases, a custom database was prepared. This database consists of in-silico translated reference genomes for K. pneumoniae (http://www.ncbi.nlm.nih.gov/genome/815?genome_assembly_id=168877) and E. coli (http://www.ncbi.nlm.nih.gov/genome/167?genome_assembly_id=161521), supplemented with a comprehensive list of beta-lactamases continued from the former Lahey database (December 2015, ftp://ftp.ncbi.nlm.nih.gov/pathogen/betalactamases/Allele-prot.fa). Searches were carried out with the following parameters: precursor mass tolerance was 0.05 Da and MS/MS tolerance 0.8 Da. Carbamidomethylcysteine was set as a fixed modification, with methionine oxidation as variable modification. Trypsin was designated as an enzyme with a maximum allowed number of missed cleavages of two. The False Discovery Rate (FDR) was set at 0.01 at the peptide level based on decoy database searches.
Results
Optimization of sample extraction
Two different sample preparation protocols were compared with respect to the overall number of protein identifications and the ease of use of the method. For this purpose, we used negative blood culture bottles spiked with different amounts of liquid broth culture of E. coli BL21(DE3) pLysS cells to mimic different bacterial densities in positive blood cultures. The number of successful protein identifications was determined at 3.0 × 107 and 3.0 × 108 CFU using a sample preparation by serum separator tubes and by differential lysis protocol using saponin. As a reference, the protocols were also applied to the bacterial suspensions used for inoculation of the blood cultures, using 1.0 × 107 CFU. Results of all analyses were searched independently against the bacterial database and, for human proteins, against the human database. Table 2 summarises the results for the proteomic comparisons of both protocols. The sample containing 3.0 × 108 CFU performed better in the saponin protocol. Since this procedure is less laborious we treated all subsequent positive blood cultures by the saponin protocol. A number of variables of the saponin lysis protocol were tested including centrifugation speed and duration, saponin concentration and number of washing steps. No significant improvements were made and the protocol therefore remained unchanged.
Blood culture collection and susceptibility testing
During a period of four months, positive blood cultures with E. coli or K. pneumoniae were collected prospectively (Table 3). In total, 170 positive blood cultures were collected. Of these, 125 (73.5%) contained E. coli and 45 (26.5%) K. pneumoniae. Following susceptibility testing of cultured isolates, 22 isolates (12.9%, 18 E. coli and 4 K. pneumoniae) were confirmed as ESBL-positive with the combination disk diffusion test.
Results from bottom-up proteomics analysis
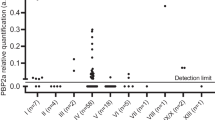
All 22 blood cultures with ESBL positive isolates were selected for proteomic analysis, as well as 44 randomly selected ESBL negative blood cultures. Preparation of the 66 blood cultures for LC-MS/MS analysis was performed blind with regards to the results of the phenotypic testing. Following LC-MS/MS analysis, the resulting spectra were searched against the in-house generated database (see materials and methods) featuring a comprehensive list of beta-lactamases as well as K. pneumoniae and E. coli proteomes. In a typical analysis of one positive blood culture bottle, 400–800 bacterial proteins were identified. Table 4 summarises the results for the phenotypically ESBL positive blood cultures (n = 22). In all results obtained by MS, the detected β-lactamase was always in the top 10% of the total number of identified bacterial proteins in a sample, sorted by identification score. In 21 out of 22 of the ESBL positive isolates a cefotaximase (CTX-M) was identified. Protein sequence coverage based on identified peptides varied from 38% to 88%. This coverage allows for the mapping of the identified cefotaximases into one of six established lineages [16], named after their archetypical enzymes. In our collection, only members of groups CTX-M-1 and CTX-M-9 were found. In one K. pneumoniae isolate (Table 4, number 15) no cefotaximase was found. A SHV type beta-lactamase was identified with 33% coverage (Fig. 1). Like with the cefotaximases, this protein was a top 10% identification among all bacterial proteins identified. From 148 ESBL-negative K. pneumonia or E. coli positive blood cultures, 44 were randomly selected and also analyzed by LC-MS/MS. In none of these samples, extended-spectrum beta-lactamases were found.

Coverage of SHV-1 sequence. Identified peptides by LC-MS/MS analysis are highlighted when they matched to the sequence of the SHV-1 beta-lactamase. The glycine at Ambler position 238 (underlined) is specific for the SHV-1 sequence, while SHV-2 type extended-spectrum beta-lactamases have a serine in this position. This peptide was not observed in LC-MS/MS analysis making it not possible to distinguish between the beta-lactamase types
Molecular characterisation of ESBL positive isolates
To confirm the identity of the ESBLs identified with LC-MS/MS based proteomics, molecular characterisation of all phenotypically ESBL positive isolates was performed (Table 4). All CTX-M identifications were verified with PCR. In isolate 15, the LC-MS/MS identified a SHV-enzyme which was confirmed by PCR as an ESBL, namely, SHV-12. Three non-ESBL SHV beta-lactamases were identified by PCR in the K. pneumoniae isolates.
Discussion
In this study we developed a novel proteomic workflow for the direct identification of ESBLs in positive blood culture bottles. To evaluate the performance of our approach, a proof-of-principle prospective study was performed in two academic hospitals. In 22 positive blood cultures with phenotypically ESBL producing E. coli or K. pneumoniae, we identified 21 isolates containing a CTX-M and one isolate containing a SHV beta-lactamase, although the latter could not unambiguously be identified because the single peptide necessary to discriminate between an ESBL and non-ESBL was not identified. In the set of positive blood cultures with ESBL-negative E. coli or K. pneumoniae, no ESBLs were identified by LC-MS/MS analysis. This demonstrates a 95% sensitivity and 100% specificity of the workflow to directly identify these beta-lactamases in positive blood cultures.
Of 170 positive blood cultures collected in two academic hospitals, 22 (12.9%) contained ESBL producing bacteria belonging to E. coli or K. pneumoniae. This percentage is higher than previously described in The Netherlands [3], with cefotaxime/ceftriaxone resistances reported to be 5% and 7% for E. coli and K. pneumoniae, respectively. However, these data were based on a larger number of laboratories, including laboratories serving non-university hospitals and general practitioners. All collected samples in our study showed full meropenem susceptibility, in agreement with the low prevalence rate of carbapenemase producing Gram-negatives in The Netherlands [3].
In the proteomic analysis of the clinical isolates, 21 out of 22 (95%) of the phenotypically ESBL-positive isolates contained a CTX-M ESBL. Cefotaximases are the most widespread ESBLs and a high representation in this study was expected [16, 17]. In our study, the CTX-M enzymes belonged to group 1 (17 out of 21; 81%) and group 9 (4 out of 21; 19%). This is in accordance with other reports [18]. Specifically, CTX-M-15 (group 1) and CTX-M-14 (group 9) are among the most prevalent enzymes [19]. Notably, in our collection there was no relation between MIC and ESBL type, especially not for ceftazidime.
Full sequence coverage is necessary to pinpoint a protein identification to a specific ESBL but in complex samples with the use of only one proteolytic enzyme, this is not feasible. Obviously, peptide fractionation or additional experiments with another proteolytic enzyme could improve the specificity of the identification. However, we opted for a simple sample preparation protocol, which is mostly constrained time-wise by the proteomic digestion step. In our approach, the sequence coverage among ESBLs in phenotypically positive isolates ranged between 38% and 88%. This coverage is in-depth enough to classify the enzymes into phylogenetic groups, such as with the cefotaximases, but single variants cannot be distinguished using this method. This is important in distinguishing beta-lactamases that have reported broad and extended-spectrum activities, based on small permutations. For example, one blood culture sample contained a SHV beta-lactamase. The sequence coverage obtained by LC-MS/MS analysis was not sufficient to distinguish between a broad and extended-spectrum beta-lactamase. The amino acid at position 238 is instrumental in cephalosporin resistance in SHV variants and the tryptic peptide covering this amino acid is therefore necessary for the unambiguous assignment of the ESBL status [20]. The corresponding tryptic peptides of the SHV-1 (broad spectrum) and SHV-2 (extended-spectrum) are TGAGER and TGASER, respectively. While the double-charged state would be within the mass range of the mass spectrometer, sensitivity in this low mass range is not optimal and short peptides can also be difficult to retain and separate in liquid chromatography. A more targeted approach might be more suitable for such specific peptides [21]. Importantly, the LC-MS/MS identified SHV beta-lactamase in isolate 15 was confirmed to be an ESBL (SHV-12) by our PCR and sequence analysis. The PCR analyses revealed three additional SHV beta-lactamases which were not identified in the proteomics analysis. Sequence analysis demonstrated that the three additional SHV beta-lactamases were non-ESBLs (SHV-1, SHV-11) and could have been missed in our proteomic analysis due to lower abundance as compared to the ESBL-SHV. Therefore, as it stands now, identifying a SHV with high expression combined with the phenotypical results indicates an ESBL-SHV, but our proteomic data was not sufficient to unambiguously draw this conclusion.
In this proof-of-principle study, ESBLs from the CTX-M group were easy to identify. PCR-based methods have been successfully applied for the identification of ESBLs in blood cultures [22, 23], and in our study, the CTX-M PCR results fully correlated with the proteomics results. The aim of our prospective study was to demonstrate the applicability in normal routine, and therefore we detected mainly CTX-M ESBL. Larger clinical sample cohorts and spiking experiments with other ESBL/carbapenemase producing bacteria in negative blood cultures are necessary to demonstrate the general applicability of our approach. Based on the results of our previous study, this workflow should also be suitable for the detection of OXA-48 and KPC beta-lactamases directly in blood cultures [11]. The sample preparation is highly similar and overall proteome and protein coverage was significantly higher using this nanoLC platform. Moreover, the mass spectrometric analysis part of our workflow can be easily exchanged for other high-end mass spectrometry analysers (such as Orbitraps) with even higher speed and sensitivities. As with all genetic methods, a positive identification does not guarantee protein expression. More sensitive proteomic analysis could therefore give some insight into our problem to detect the additional non-ESBL lactamases with our proteomics workflow.
Apart from genetic tests, there are alternative methods to detect the presence of ESBLs in blood culture bottles. Oviaño et al. monitored ESBL activity directly from blood cultures using MALDI-TOF MS by measuring antibiotic hydrolysis [24]. Reported sensitivity and specificity are high, suggesting that such an approach can be used as an alternative to traditional susceptibility testing. Moreover, hydrolysis based assays using reporter molecules are mentioned in literature and available as commercial kits [25, 26]. Even though hydrolysis based tests are useful, interpretation can be difficult in case of enzymes with a lower activity, and they provide no insight in the identity of the ESBL. In comparison with genetic and hydrolysis based methods, our workflow allows the direct identification of the enzyme responsible, providing molecular information about the phenotype.
Overall, in this proof-of-principle prospective study we demonstrate the direct identification of an ESBL in all blood cultures that contained bacteria positive for a CTX-M type ESBL. The method is specific enough to recognise specific groups of CTX-M ESBL. To improve on this proof-of-principle study in the future a number of aspects need further exploration. Among these, shortening the time-to-report and automation of the procedure are among the most critical [27, 28]. With this in mind, the developed platform can be used in the future for the direct identification of expressed beta-lactamases in blood cultures which provides detailed insight into the antibiotic resistance mechanism.
References
Souverein D, Euser SM, Herpers BL, Diederen B, Houtman P, van Seventer M, van Ess I, Kluytmans J, Rossen JW, Den Boer JW (2016) Prevalence, risk factors and molecular epidemiology of highly resistant gram negative rods in hospitalized patients in the Dutch region Kennemerland. Antimicrob Resist Infect Control 5:8. doi:10.1186/s13756-016-0107-6
Tacconelli E, Cataldo MA, Dancer SJ, De Angelis G, Falcone M, Frank U, Kahlmeter G, Pan A, Petrosillo N, Rodriguez-Bano J, Singh N, Venditti M, Yokoe DS, Cookson B (2014) ESCMID guidelines for the management of the infection control measures to reduce transmission of multidrug-resistant gram-negative bacteria in hospitalized patients. Clin Microbiol Infect 20(Suppl 1):1–55. doi:10.1111/1469-0691.12427
de Greeff SC, Mouton JW, Schoffelen AF (2016) NethMap 2016: Consumption of antimicrobial agents and antimicrobial resistance among medically important bacteria in The Netherlands in 2015. RIVM, Bilthoven, The Netherlands
Patel R (2015) MALDI-TOF MS for the diagnosis of infectious diseases. Clin Chem 61(1):100–111. doi:10.1373/clinchem.2014.221770
Tomazella GG, Risberg K, Mylvaganam H, Lindemann PC, Thiede B, Souza GA, Wiker HG (2012) Proteomic analysis of a multi-resistant clinical Escherichia coli isolate of unknown genomic background. J Proteome 75(6):1830–1837. doi:10.1016/j.jprot.2011.12.024
Pinto L, Poeta P, Vieira S, Caleja C, Radhouani H, Carvalho C, Vieira-Pinto M, Themudo P, Torres C, Vitorino R, Domingues P, Igrejas G (2010) Genomic and proteomic evaluation of antibiotic resistance in salmonella strains. J Proteome 73(8):1535–1541. doi:10.1016/j.jprot.2010.03.009
Maravic A, Cvjetan S, Konta M, Ladouce R, Martin FA (2016) Proteomic response of beta-lactamases-producing Enterobacter cloacae Complex strain to cefotaxime-induced stress. Pathog Dis 74(5). doi:10.1093/femspd/ftw045
Trip H, Mende K, Majchrzykiewicz-Koehorst JA, Sedee NJ, Hulst AG, Jansen HJ, Murray CK, Paauw A (2015) Simultaneous identification of multiple beta-lactamases in Acinetobacter baumannii in relation to carbapenem and ceftazidime resistance, using liquid chromatography-tandem mass spectrometry. J Clin Microbiol 53(6):1927–1930. doi:10.1128/jcm.00620-15
Chang CJ, Lin JH, Chang KC, Lai MJ, Rohini R, Hu A (2013) Diagnosis of beta-lactam resistance in Acinetobacter Baumannii using shotgun proteomics and LC-nano-electrospray ionization ion trap mass spectrometry. Anal Chem 85(5):2802–2808. doi:10.1021/ac303326a
Osei Sekyere J, Govinden U, Essack SY (2015) Review of established and innovative detection methods for carbapenemase-producing gram-negative bacteria. J Appl Microbiol 119(5):1219–1233. doi:10.1111/jam.12918
Fleurbaaij F, Heemskerk AA, Russcher A, Klychnikov OI, Deelder AM, Mayboroda OA, Kuijper EJ, van Leeuwen HC, Hensbergen PJ (2014) Capillary-electrophoresis mass spectrometry for the detection of carbapenemases in (multi-)drug-resistant gram-negative bacteria. Anal Chem 86(18):9154–9161. doi:10.1021/ac502049p
Bakthavatchalam YD, Anandan S, Veeraraghavan B (2016) Laboratory detection and clinical implication of oxacillinase-48 like carbapenemase: the hidden threat. J Glob Infect Dis 8(1):41–50. doi:10.4103/0974-777x.176149
Ferroni A, Suarez S, Beretti JL, Dauphin B, Bille E, Meyer J, Bougnoux ME, Alanio A, Berche P, Nassif X (2010) Real-time identification of bacteria and Candida species in positive blood culture broths by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 48(5):1542–1548. doi:10.1128/jcm.02485-09
NVMM (2013) Laboratory detection of highly resistant microorganisms (HRMO). http://www.nvmm.nl/richtlijnen/hrmo-laboratory-detection-highly-resistant-microorganisms (in Dutch). Accessed 04 April 2017
Paltansing S, Kraakman ME, Ras JM, Wessels E, Bernards AT (2013) Characterization of fluoroquinolone and cephalosporin resistance mechanisms in Enterobacteriaceae isolated in a Dutch teaching hospital reveals the presence of an Escherichia coli ST131 clone with a specific mutation in parE. J Antimicrob Chemother 68(1):40–45. doi:10.1093/jac/dks365
D’Andrea MM, Arena F, Pallecchi L, Rossolini GM (2013) CTX-M-type beta-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol 303(6–7):305–317. doi:10.1016/j.ijmm.2013.02.008
Naseer U, Sundsfjord A (2011) The CTX-M conundrum: dissemination of plasmids and Escherichia coli clones. Microb Drug Resist 17(1):83–97. doi:10.1089/mdr.2010.0132
Zhao WH, Hu ZQ (2013) Epidemiology and genetics of CTX-M extended-spectrum beta-lactamases in gram-negative bacteria. Crit Rev Microbiol 39(1):79–101. doi:10.3109/1040841x.2012.691460
Cantón R, Novais A, Valverde A, Machado E, Peixe L, Baquero F, Coque TM (2008) Prevalence and spread of extended-spectrum β-lactamase-producing Enterobacteriaceae in Europe. Clinical Microbiology and infection 14. Supplement 1:144–153. doi:10.1111/j.1469-0691.2007.01850.x
Hujer AM, Hujer KM, Helfand MS, Anderson VE, Bonomo RA (2002) Amino acid substitutions at ambler position Gly238 in the SHV-1 beta-lactamase: exploring sequence requirements for resistance to penicillins and cephalosporins. Antimicrob Agents Chemother 46(12):3971–3977
Charretier Y, Dauwalder O, Franceschi C, Degout-Charmette E, Zambardi G, Cecchini T, Bardet C, Lacoux X, Dufour P, Veron L, Rostaing H, Lanet V, Fortin T, Beaulieu C, Perrot N, Dechaume D, Pons S, Girard V, Salvador A, Durand G, Mallard F, Theretz A, Broyer P, Chatellier S, Gervasi G, Van Nuenen M, Roitsch CA, Van Belkum A, Lemoine J, Vandenesch F, Charrier JP (2015) Rapid bacterial identification, resistance, virulence and type profiling using selected reaction monitoring mass spectrometry. Sci Rep 5:13944. doi:10.1038/srep13944
Opota O, Croxatto A, Prod’hom G, Greub G (2015) Blood culture-based diagnosis of bacteraemia: state of the art. Clin Microbiol Infect 21(4):313–322. doi:10.1016/j.cmi.2015.01.003
Fishbain JT, Sinyavskiy O, Riederer K, Hujer AM, Bonomo RA (2012) Detection of extended-spectrum beta-lactamase and Klebsiella pneumoniae Carbapenemase genes directly from blood cultures by use of a nucleic acid microarray. J Clin Microbiol 50(9):2901–2904. doi:10.1128/jcm.01023-12
Oviano M, Fernandez B, Fernandez A, Barba MJ, Mourino C, Bou G (2014) Rapid detection of enterobacteriaceae producing extended spectrum beta-lactamases directly from positive blood cultures by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Clin Microbiol Infect 20(11):1146–1157. doi:10.1111/1469-0691.12729
Nordmann P, Dortet L, Poirel L (2012) Rapid detection of extended-Spectrum-β-lactamase-producing Enterobacteriaceae. J Clin Microbiol 50(9):3016–3022. doi:10.1128/jcm.00859-12
Jain S, Andrews J, Fraise A, Brenwald N (2007) Rapid detection of extended-spectrum beta-lactamase-producing gram-negative bacilli in blood cultures. J Antimicrob Chemother 60(3):652–654. doi:10.1093/jac/dkm256
Riley Nicholas M, Hebert Alexander S, Coon Joshua J Proteomics moves into the fast lane. Cell Systems 2(3):142–143. doi:10.1016/j.cels.2016.03.002
Regnier FE, Kim J (2014) Accelerating trypsin digestion: the immobilized enzyme reactor. Bioanalysis 6(19):2685–2698. doi:10.4155/bio.14.216
Acknowledgements
This research was financially supported by The Netherlands organisation of scientific research (NWO, ZonMW grant number 50-51700-98-142).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Not applicable (as discussed with the institutional medical ethical committee).
Additional information
Paul J. Hensbergen and Ed J. Kuijper share senior authorship.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fleurbaaij, F., Goessens, W., van Leeuwen, H.C. et al. Direct detection of extended-spectrum beta-lactamases (CTX-M) from blood cultures by LC-MS/MS bottom-up proteomics. Eur J Clin Microbiol Infect Dis 36, 1621–1628 (2017). https://doi.org/10.1007/s10096-017-2975-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-017-2975-y