
Abstract
In general, concepts such as chemical equilibria and classical analytical chemistry, as well as instrumental methods such as potentiometry and flow injection analysis (FIA), are taught separately in the curricula of chemistry-related degree programs. However, FIA provides an opportunity to connect all of these important topics. This article shows how simple experiments can be used to link the fundamentals of FIA and potentiometry to the fundamentals of chemical equilibria and teach them in a sustainable way. The experiments were designed for undergraduate environmental science students with basic knowledge of chemical equilibria and classical analytical chemistry and minimal knowledge of electrochemistry. However, they can also be integrated into various courses on instrumental analysis in chemistry-related degree programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flow-based analytical techniques with injection of the test sample have a high value in instrumental analysis, as they offer the possibility to automate and minimize the analysis. These are relevant topics, since the requirements for minimizing the consumption of samples, reagents, and time, as well as minimizing the amount of waste, have to be taken into account for improving analytical methods used in lab courses of students, as well as in practice. A good overview of the basic principles and most relevant developments in flow-based analytical methods has been given by Timofeeva et al. [1]. However, educational papers on flow injection analysis are still rare, although experiments can be performed with a fairly simple basic configuration [2]. Electrochemical and especially potentiometric detectors can greatly simplify the setup. Understanding of sensors and especially potentiometric sensors is a prerequisite [3].
In this manuscript, it is shown that simple experiments can be performed to demonstrate the principles of flow injection titration with a potentiometric detector for acid–base, complexometric, and redox titrations. In flow injection titrations, the product of the reaction between a discrete volume of sample solution injected into a carrier reagent stream and the carrier reagent solution is monitored with a flow-through detector. The principles of the processes occurring during the residence time of the injected solution in the carrier solution (convection, diffusion, reactions at the front and back sites of the sample zone [4]) and several methodologies of flow injection titrations (e.g., flow injection titrations with or without a mixing chamber [5,6,7]) are part of a theoretical lesson.
It is important to consider the differences between a purely physical dispersion (no chemical reaction between sample and carrier solution) and a physical dispersion with chemical reactions (a flow injection titration). In the first case (e.g., pH measurements by injecting the sample into a KCl electrolyte solution), the peak height can be used to calibrate the system to illustrate that the potentiometric detector signal is proportional to the pH of the solution. In case of FIA titrations, the peak width as well as the peak area can be used for quantification. In classical solution titrations (volumetric titrations), a known volume of sample is titrated by a measured volume of titrator of known concentration. When potentiometry is used to indicate the titration, the titration curve can be plotted as pH versus added volume of titrator. The end point is indicated by an inflection point and the steepest pH rise. In FIA titrations, the analyte reacts gradually in the dispersion zone with the reagent carrier solution (titrator). Three segments can be distinguished on both sides of the peak: (i) the titrator is present in excess; (ii) the amounts of analyte and titrator are equal; and (iii) the analyte is present in excess. This is only the case if the dispersion is larger than 1.0 and the analyte concentration exceeds that of the titrator. If the analyte concentration is less than that of the titrator, inflection points still occur on both sides of the peak. Hence, in FIA titration the peak, width and the peak area are functions of the analyte concentration.
Laboratory sessions consist of three experiments on real samples, which the students can provide themselves. The students have to prepare a protocol for the experiments. These protocols also include a theoretical part, in which various questions about the experiment and the reactions and transport processes taking place have to be answered. The proposed experiments allow the exploration of the phenomena and concepts of FIA (e.g., peak formation, dispersion, detectors, interpretation of the obtained signals) and of potentiometry (potential formation, Nernst equation, reference electrodes, and indicator electrodes). In addition, concepts and routines of analytical chemistry are taught (evaluation of raw data, preparation, and evaluation of calibration curves, etc.).
Experiments
FIA configuration
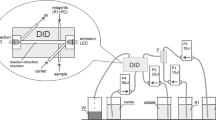
A single-line FIA configuration is used for all experiments. The carrier stream is propelled using a peristaltic pump (ISMATECH, Switzerland). A 6-port-valve (Rheodyne, USA) is used to inject the sample into the carrier stream. The setup of the home-made detector cell is shown in Fig. 1. The indicator electrode for acid–base and complexometric titrations consists of a graphite/quinhydrone/silicon composite electrode as described in [8]. For redox titrations, the electrode consists of graphite/silicon composite only. The main advantages of these electrodes used are the mechanical flexibility due to the silicon content and the fact that no further sealing of the flow system is necessary. The flow channel can be pierced through the electrode with a canula of suitable size. A conventional reference electrode (in our case a silver/silver chloride electrode with 3 M KCl as internal electrolyte) is connected to the detector cell via a salt bridge (a tube filled with a gel made of saturated KCl and agarose) [9]. Chronopotentiometric measurements are performed using an AUTOLAB with a PSTAT10 (Ecochemie, The Netherlands); however, it is possible to replace the AUTOLAB by other commercial potentiometric measuring devices. Data acquisition rate is chosen as 5 points per second.

Schematic description of the detector cell: A indicator electrode, B and C plexiglass plates, D screws to fix the plates, E flow channel (diameter 0.5 mm), F tube fittings, G copper wire for electrical contact, H gel filled salt bridge
Titratable acidity and pH of wine
The pH and titratable acidity of wine play an important role in oenology, as both parameters influence the taste, color, and microbiological stability of the wine. One method to determine the pH and titratable acidity of wine has been developed in our lab in 2013 (experimental details are described in [10]) and was modified as a student experiment. In our experience, the experiment is very attractive for students because real samples are examined, which they can provide themselves. The theoretical part includes the following questions:
-
(i)
What are the main acids to be expected in wine, and what are the expected concentrations of these acids?
-
(ii)
What are the chemical properties of these acids, and what are their acidity constants? This leads to the question of how to titrate these acids in a classical way and where to expect the equivalence points. Here, students need to reactivate their knowledge of acid–base equilibria [11], classical acid–base titrations, and indicators used for the latter [12].
-
(iii)
What are the standard methods for determining the titratable acidity of wine samples in the EU and the USA?
-
(iv)
A composite electrode made of graphite/quinhydrone/silicone is used for acid–base titration. What is the response mechanism of the pH-dependent redox system quinhydrone (see [11] p.150–156)? What would happen if there is another redox-active system in the analyte solution? The last question is important for calibration for the determination of titratable acidity. This is because wine is a complex matrix that contains additional pH-dependent redox systems that may interfere with the detector response [10]. Therefore, the matrix of the standard solutions used for calibration must be similar to the wine matrix. The solvent used for all calibration solutions and the solvent for the dilution of all wine samples is a wine (red wine or white wine depending on the samples to be analyzed) titrated with 4 mol L−1 sodium hydroxide to the equivalence point (pH ~ 7.5, measured with a conventional glass electrode using a pH meter set Qph 70, (VWR international GmbH, Germany).
-
(v)
What processes occur at the front and back sites of the peak (i.e., the two dispersion zones) when an alkaline solution is used as the carrier solution?
Students have to prepare appropriate calibration solutions and dilutions of wine samples. Since malic acid (2-hydroxybutanedioic acid) is one of the major non-volatile acids in wine, DL-malic acid is used for calibration. Appropriate means here that the expected sample concentration lies in the middle of the calibration curve. Since the concentrations of titratable protons in wines are usually in the range of 60 to 90 mmol L−1, the concentration range for the calibration can be kept quite narrow, because then a linear calibration curve is obtained when peak areas are used for evaluation. When the range is extended, non-linear calibration curves result (cf. [13]). For the calibration curve shown in Fig. 2, the concentrations of titratable protons are in the range of 1 to 25 mmol L−1, so that the wine samples have to be diluted by a factor of five or ten. The wine samples need to be purged with nitrogen for approximately 10 min in order to eliminate carbon dioxide before dilution.

Typical calibration curve (peak area vs. concentration of hydronium ions; white wine adjusted to pH 7.5 served as the solvent)
For pH measurements, a carrier solution of 0.01 M hydrochloric acid and 0.01 M potassium chloride is used. Hydrochloric acid is used at this high concentration to ensure that the baseline potential was greater than the peak potential of the samples. Britton-Robinson buffer solutions in a pH range from 2 to 8 are used for calibration [14]. The FIA setting remains unchanged. Here, peak heights are used for evaluation to make it clear that this is not a flow injection titration. Students need to discuss the sub-Nernstian slope (small sample volume and rapid determination do not lead to steady-state signals) and the deviation from linearity in more alkaline solutions due to the properties of the quinhydrone redox system [15].
Determination of water hardness
Since the determination of calcium and magnesium ions in aqueous solutions is one of the most frequently performed analyses, and classical titrations of these ions with EDTA salts are standard undergraduate experiments, we decided to adapt a FIA titration experiment for determination of these ions [8]. The method is based on the principle that protons are released when the disodium or trisodium salt of EDTA is used for complexation. Thus, the potential at the graphite/quinhydrone/silicon composite electrode changes when a water sample is injected into a carrier stream of disodium or trisodium salt of EDTA. Complexation of calcium and magnesium ions with EDTA anions gradually takes place at the front and back sides of the peak, resulting in proton release. The experiment consists of two parts:
-
(i)
Students investigate the effect of the nature of the carrier solution (the pH values of the solutions of disodium or trisodium salt of EDTA are different) on the peak shape.
This experiment reactivates the basics of complexometric equilibria (see Chapter 4 in [11]). Two carrier solutions are used for this experiment. Carrier solution 1 is prepared by dissolving 0.01 mol L−1 of the disodium salt of EDTA, and carrier solution 2 is prepared by dissolving a Titrisol ampoule (Merck, Germany) in 5 L ultrapure water to obtain a 0.02 mol L−1 solution of the trisodium salt. Potassium chloride is added to both carrier solutions at a concentration of 0.01 mol L−1 to achieve asufficiently high conductivity to give a smooth base line. First, the students measure the pH of the two carrier solutions. Carrier solution 1 has a pH of 4.5, and carrier solution 2 has a pH of 9.1. If the pH of the sample is close to 7.0, negative peaks are observed in the case of carrier solution 1, and the peak shape can be discussed by the students (a detailed discussion of the explanation of different peak shapes is provided in [8]). The conclusion is that carrier solution 2 will be used for further experiments.
-
(ii)
The water hardness of different real samples is determined by the students. For this, FIA titrations are performed in two steps.
Students have to prepare appropriate calibration solutions. Since the water hardness and thus the calcium and magnesium ion content in natural waters can vary greatly, a concentration range of three to four concentration decades is selected for calibration. To determine the sum of calcium and magnesium ions, the system is calibrated with equimolar calcium and magnesium ion solutions. All standard solutions have to contain 0.01 mol L−1 potassium chloride and need to be adjusted to pH 6, because in [8] it has been demonstrated that the influence of the pH of the sample solution is negligible in a pH range from 4.5 to 7.0. The reason for this is that two effects affect the peak area: Since a potentiometric pH sensor is used, the peak area increases with decreasing pH, and at the same time, the effective stability constants of the formed calcium and magnesium EDTA complexes decrease. This also reduces the number of released protons. Since the pH value of natural water is around seven, samples can be injected without pre-treatment. It is worth to mention that the calibration curve (peak area vs. concentration) is not linear (see Fig. 3; a detailed discussion of reasons why the calibration curve follows a sigmoidal growth function is also provided in [8]).

Dependence of peak areas on the concentration of the sum of calcium and magnesium ions (equimolar solutions)
The real water samples are injected in the FIA system without further pre-treatment. To determine the calcium ion concentration only, the system is calibrated with calcium ion standard solutions. In real water samples, magnesium ions are precipitated by adding 5 mL of 1 mol L−1 NaOH to 40 mL sample and adjusting the volume to 50 mL in a measuring flask. Here, students can be reminded to the pH dependence of oxides and hydroxides [16]. To get rid of the solid magnesium hydroxide, the solution is filtered through a syringe filter. To 25 mL of the filtrate, hydrochloric acid is added to adjust the pH of 6, and the volume is filled of to 50 mL in a measuring flask. This solution is injected into the carrier solution in the FIA system.
Redox titrations
For redox titrations, the composite electrode consists only of a graphite/silicon layer. This means that only the electrode layer in the middle of the detector cell has to be replaced, and then the cell can be used in the same arrangement as for the other experiments. As a student experiment, we have chosen the cerimetric determination of ascorbic acid (vitamin C) in a commercial vitamin C preparation. A 5∙10−3 mol L−1 solution of cerium (IV) sulfate is used as the carrier solution. It is important to note that for all experiments described here, a 0.5 M solution of sulfuric acid is used as the solvent. Prior to the experiment, students should review the basics of redox equilibria (see Chapter 6 in [11]) and the structure and redox properties of ascorbic acid.
For calibration, a stock solution of ascorbic acid of 0.1 M in sulfuric acid and then suitable calibration solutions ranging from 0.1 to 0.001 mol L−1 and a blank solution (0.5 M sulfuric acid) are freshly prepared by the students and injected into the carrier stream. The sample is prepared by dissolving one tablet of the vitamin C preparation (e.g., Abtei OP Pharma GmbH, Germany, vitamin C + zinc, 250 mg ascorbic acid per tablet) in 0.5 M sulfuric acid in a 250-mL flask.
A typical potential-time curve is given in Fig. 4. For calibration, the peak area is plotted versus the log concentration of the calibration solutions, as shown in Fig. 5.

Typical potential-time curve for the injection of two different concentrations of ascorbic acid solutions into the carrier solution of cerium (IV) sulfate

Typical calibration curve for the cerimetric determination of ascorbic acid. Peak areas are plotted versus log concentration of ascorbic acid
We have chosen cerium sulfate solution as the titrator because the oxidation reaction is fast and the potentiometric response is excellent. The volume of produced waste solution is very small compared to classical titrations. Nevertheless, it would be nice when readers of this paper will—perhaps with their students—search for alternative and less harmful titrators.
Conclusions
The proposed FIA setup is very robust and flexible, because the electrode layer can be easily replaced by the students. In lab courses at our university, the setup has been proven to be of great value to help students understand classical chemical equilibria and instrumental methods like potentiometry and flow injection analysis. The analysis of real samples leads to a more interesting lesson, which stimulates the students’ curiosity.
References
Timofeeva I, Nugbienyo L, Pochivalov A, Vakh C, Shishov A, Bulatov A (2021) Flow-based methods and their applications in chemical analysis. ChemTexts 7:24. https://doi.org/10.1007/s40828-021-00149-8
Augustini D, Berganini MF, Marcolino-Junior LH (2018) Simple and inexpensive microfluidic thread based device for teaching microflow injection analysis and electrochemistry. J Chem Educ 95:1411. https://doi.org/10.1021/acs.jchemed.8b00211
Gründler P (2017) Chemical sensors. ChemTexts 3:16. https://doi.org/10.1007/s40828-017-0052-x
Karlberg B, Pacey GE (1989) Flow injection analysis. A practical guide. In: Techniques and instrumentation in analytical chemistry, Vol. 10. Elsevier, Amsterdam
Růžička J, Hansen EH, Mosbæk H (1977) Flow injection analysis. Anal Chim Acta 92:235. https://doi.org/10.1016/S0003-2670(01)93497-X
Tyson JF (1987) The principles of flow injection titration. Anal Proc 24:362. https://doi.org/10.1039/AP9872400359
Ruzicka J (2019) Flow injection analysis: tutorial & news on flow based micro analytical techniques. https://flowinjectionturorial.com. Accessed 21 June 2023
Vahl K, Kahlert H, Scholz F (2010) Rapid automatic determination of calcium and magnesium in aqueous solutions by FIA using potentiometric detection. Electroanal 22:2172. https://doi.org/10.1002/elan.201000146
Scholz F (2013) Common types of salt bridges and diaphragms. Pp 54, In: Handbook of reference electrodes. A. Lewenstam, G. Inzelt, F. Scholz (editors), Springer, Berlin, ISBN 978-3-642-36187-6, eBook ISBN 978-3-642-36188-3. https://doi.org/10.1007/978-3-642-36188-3
Vahl K, Kahlert H, von Mühlen L, Albrecht A, Meyer G, Behnert J (2013) Determination of the titratable acidity and the pH of wine based on potentiometric flow injection analysis. Talanta 111:134. https://doi.org/10.1016/j.talanta.2013.02.057
Scholz F, Kahlert H (2019) Chemical equilibria in analytical chemistry. The theory of acid-base, complex, precipitation, and redox equilibria. Springer Nature, Cham, ISBN 978-3-030-17179-7 ISBN 978-3-030-17180-3 (eBook) https://doi.org/10.1007/978-3-030-17180-3
Kahlert H, Meyer G, Albrecht A (2016) Colour maps of acid–base titrations with colour indicators: how to choose the appropriate indicator and how to estimate the systematic titration errors. ChemTexts 2:7. https://doi.org/10.1007/s40828-016-0026-4
Kahlert H, Pörksen JR, Behnert J, Scholz F (2005) FIA acid–base titrations with a new flow-through pH detector. Anal Bioanal Chem 382:1981. https://doi.org/10.1007/s00216-005-3379-x
Britton HTH, Robinson RA (1931) Universal buffer solutions and the dissociation constant of veronal. J Chem Soc. https://doi.org/10.1039/JR9310001456
Kahlert H (2010) Potentiometry. In: Scholz F (ed) Electroanalytical methods, 2nd edn. Springer, Berlin, Heidelberg, p 240
Scholz F, Kahlert H (2015) The calculation of the solubility of metal hydroxides, oxide-hydroxides, and oxides, and their visualisation in logarithmic diagrams. ChemTexts 1:7. https://doi.org/10.1007/s40828-015-0006-0
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kahlert, H., Meyer, G. & Albrecht, A. Potentiometry, chemical equilibria, and flow injection analysis—three important topics taught with one experimental setup. J Solid State Electrochem 28, 1191–1197 (2024). https://doi.org/10.1007/s10008-023-05598-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-023-05598-4