
Abstract
Context
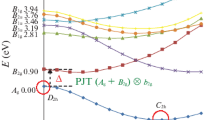
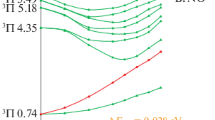
The Pseudo Jahn- Teller effect is a significant tool for evaluating molecular distortion and symmetry breaking. The PJT effect associated with NBO analysis can be a powerful method for studying the structural properties variations arising from D2h → C2h distortions. The theoretical studies on Si2X4+ and Ge2X4+ radical cations have been rare. The calculations have shown that C2h non-planar structures are more stable than planar structures with D2h symmetry. The \(({B}_{3u}+{B}_{1u})\otimes {b}_{2g}\) PJTE problem of M2X4+ compounds is a result of the coupling between the ground B3u state and the exited B1u state in the Qb2g direction causes. Also, the difference in M and X atoms can affect the PJT instability of compounds. The findings of this work show that the energy gap between the ground and excited states that have D2h symmetry decreases from M2Cl4+ to M2I4+ and increases from Si2X4+ to Ge2X4+. In fact, there is a significant relationship between instability of high-symmetry configurations, geometric parameters, electron delocalization, chemical hardness, electronegativity, electrophilicity index, and PJT stabilization energies. These results may serve to evaluate the distortion of similar systems.
Methods
The structures of Si2X4+ and Ge2X4+ are optimized by LC˗BLYP, M06˗2X, and B3LYP methods with def2˗TZVPP basis set in GAMESS software. The details of the excited states of compounds are studied by the TD-DFT method. NBO analysis for planar and non˗planar structures is carried out at B3LYP/def2˗TZVPP level by the NBO 5. G program that demonstrates HOMO, LUMO, ED, bonding and antibonding orbital occupancies, bond order, and E2.






Similar content being viewed by others

Abbreviations
- Pseudo Jahn–Teller:
-
(PJT)
- Natural Bond Orbital:
-
(NBO)
- Electron delocalization:
-
(ED)
- M2X4 + :
-
(M = Si, Ge, X = Cl, Br, I)
- Density functional theory:
-
(DFT)
- Adiabatic potential energy surface:
-
(APES)
- Time-dependent density functional theory:
-
(TD-DFT)
- The PJT stabilization energies:
-
(ΔEPJT)
- The highest occupied molecular orbital:
-
(HOMO)
- The lowest unoccupied molecular orbital:
-
(LUMO)
- Localized molecular orbitals:
-
(LMO)
- Canonical molecular orbitals:
-
(CMO)
- The stabilization energy:
-
(E2)
- Wiberg bond index matrix:
-
(WBI)
- The natural atomic charges:
-
(NAC)
- The natural atomic orbitals:
-
(NAO)
- Hardness:
-
(η)
- Electronegativity:
-
(χ)
- Chemical potential:
-
(μ)
- Electrophilicity index:
-
(ω)
- Electron donating power:
-
(ω−)
- Electron accepting power:
-
(ω+)
- Net electrophilicity:
-
(∆ω±)
References
D. Nori-Shargh, S. N. Mousavi and J. E. Boggs, J. Phys. Chem. A, 2013, 117, 1621 Pseudo Jahn−Teller Effect and Natural Bond Orbital Analysis of Structural Properties of Tetrahydridodimetallenes M2H4, (M = Si, Ge,and Sn) https://doi.org/10.1021/jp310389q
Inoue Sh, Ichinohe M, Sekiguchi A (2008) The Isolable Cation Radical of Disilene: Synthesis, Characterization, and a Reversible One-Electron Redox System. J AM CHEM SOC 130:6078–6079
Schorpp M, Heizmann T, Schmucker RS, Weber S, Krossing I (2020) Synthesis and Application of a Perfluorinated Ammoniumyl Radical Cation as a Very Strong Deelectronator. Angew Chem Int Ed 59:9453–9459
Rivard E (2016) Group 14 inorganic hydrocarbon analogues. Chem Soc Rev 45:989–1003
Krogh-Jespersen K (1982) Geometries and relative energies of singlet silylsilylene and singlet disilene. J Phys Chem 86(9):1492–1495
Power PP (2020) An Update on Multiple Bonding between Heavier Main Group Elements: The Importance of Pauli Repulsion, Charge-Shift Character, and London Dispersion Force Effects. Organometallics 39(23):4127–4138
Kouchakzadeh G, Nori-Shargh D (2015) Symmetry breaking in the planar configurations of disilicon tetrahalides: Pseudo-Jahn–Teller effect parameters, hardness and electronegativity. Phys Chem Chem Phys 17:29251–29261
Bersuker IB (2006) The Jahn-Teller Effect. Cambridge University Press, New York
Ilkhani AR, Monajjemi M (2015) The pseudo Jahn-Teller effect of puckering in pentatomic unsaturated rings C4AE5, A = N, P, As, E = H, F. Cl Comput Theor Chem 1074:19–25
Liu Y, Wang Y, Bersuker IB (2016) Geometry, Electronic Structure, and Pseudo Jahn-Teller Effect in Tetrasilacyclobutadiene Analogues. Sci Rep 6:23315
Bersuker IB, Polinger V (2020) Perovskite Crystals: Unique Pseudo-Jahn–Teller Origin of Ferroelectricity, Multiferroicity, Permittivity, Flexoelectricity, and Polar Nanoregions. Condens Matter 5(4):68
Kanakati AK, Rani VJ, Mahapatra. (2022) The Jahn-Teller and pseudo-Jahn–Teller effects in the propyne radical cation. Phys Chem Chem Phys 24:16522–16537
Garcia-Fernandez P, Liu Y, Bersuker IB, Boggs JE (2011) Pseudo Jahn-Teller origin of cis-trans and other conformational changes. The role of double bonds. Phys Chem Chem Phys 13:3502–3513
Nori-Shargh D, Weinhold F (2018) Natural Bond Orbital Theory of Pseudo-Jahn-Teller effects. J Phys Chem A 122:4490–4498
Kouchakzadeh G (2021) The Connection between Pseudo-Jahn–Teller-Effect and Electron Delocalization for Proving the Origin of Equilibrium Geometry of Nitrosyl Halides (Halides = Cl, Br, and I). Russ J Phys Chem A 95(2):332–342
Trinquier G, Barthelat JC (1990) Structures of X2F4, from carbon to lead. Unsaturation through fluorine bridges in Group 14. J Am Chem Soc 112:9121–9130
Li Q, Li G, Xu W, Xie Y, Schaefer HF III (2002) Molecules for Materials: Structures, Thermochemistry, and Electron Affinities of the Digermanium Fluorides Ge2Fn/Ge2F (n=1–6): A Wealth of Unusual Structures. Chem Phys Chem 3(2):179–194
Liang C, Allen LC (1990) Group IV double bonds: shape deformation and substituent effects. J Am Chem Soc 112(3):1039–1041
Allen TL, Fink WH, Power pp. (2000) Theoretical studies of multiple bonds in gallium–gallium and germanium–germanium compounds. J Chem Soc Dalton Trans 3:407–412
Kouchakzadeh G, Jamehbozorgi S (2019) The Role of Pseudo Jahn-Teller Effect in Geometry and Electronic Parameters of Tetrahalodigermene Ge2X4 (X= Cl, Br, I). Russ J Phys Chem A 93(7):1297–1304
West R (1987) Chemistry of the Silicon-Silicon Double Bond. Angew Chem Int Ed Engl 26(12):1201–1211
Lalov AV, Boganov SE, Faustov VI, Egorov MP, Nefedov OM (2003) Experimental and quantum-chemical study of complexation of carbene analogs with dinitrogen. Direct IR-spectroscopic observation of Cl2Si·N2 complexes in low-temperature argon-nitrogen matrices. Russ Chem Bull 52:526–538
Karni M, Apeloig Y (1990) Substituent effects on the geometries and energies of the silicon-silicon double bond. J Am Chem Soc 112(23):8589–8590
Mondal KC, Dittrich B, Maity B, Koley D, Roesky HW (2014) Cyclic Alkyl(amino) Carbene Stabilized Biradical of Disilicontetrachloride. J Am Chem Soc 136(27):9568–9571
Varga Z, Hargittai M (2013) Group 14 structural variations: perhalo derivatives of the ‘“dimetallenes”’: dicarbenes, disilenes, digermenes, distannenes, and diplumbenes. Struct Chem 24:837–850
Timms PL, Kent RA, Ehlert TC, Margrave JL (1965) Silicon-Fluorine Chemistry. I. Silicon Difluoride and the Perfluorosilanes. J Am Chem Soc 87(13):2824–2828
Marsmann HC, Raml W, Hengge E (1980) 29Si-Kernresonanzmessungen an Polysilanen. 2. Isotetrasilane / 29Si NMR Measurements on Polysilanes. 2. Isotetrasilanes. Z Naturforsch B 35(12):1541–1547
Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K (2004) A long-range-corrected time-dependent density functional theory. J Chem Phys 120(18):8425–8433
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120:215–241
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305
Weigend F (2006) Accurate Coulomb-fitting basis sets for H to Rn. Phys Chem Chem Phys 8:1057–1065
Gordon MS, Schmidt MW (2005) Advances in Electronic Structure Theory: GAMESS a Decade Later. In: Dykstra CE, Frenking G, Kim KS, Scuseria GE (eds) Theory and Applications of Computational Chemistry: The First Forty Years. Elsevier, Amsterdam, pp 1167–1189
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14(11):1347–1363
Assadi M H N, Hanaor D (2013) Theoretical study on copper’s energetics and magnetism in TiO2 polymorphs. J Appl Phys 113(23). https://doi.org/10.1063/1.4811539
Sitkiewicz SP, Zaleśny R, Ramos-Cordoba E, Luis JM, Matito E (2022) How reliable are modern density functional approximations to simulate vibrational spectroscopies? J Phys Chem Lett 13(25):5963–5968
Levine, I N (2014) Quantum Chemistry. Pearson
Dokmaisrijan S, Kungwan N (2019) LC-BLYP Calculations of the Structures and Photophysical Properties of [1,3]Thiazolo[4,5-b]pyrazine Derivatives in Cyclohexane and Methanol. Journal of Physical Chemistry A 123(50):10685–10693. https://doi.org/10.1021/acs.jpca.9b09074
Lu L (2015) Can B3LYP be improved by optimization of the proportions of exchange and correlation functionals? Int J Quantum Chem 115(8):502–509
Kodikara MS, Stranger R, Humphrey MG (2018) Long-Range corrected DFT calculations of first hyperpolarizabilities and excitation energies of metal alkynyl complexes. Chem Phys Chem 19(12):1537–1546
Foresman JB, Frisch A (2015) Exploring Chemistry with Electronic Structure Methods, 3rd ed. (Gaussian, Wallingford, CT)
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F (2004) NBO Version 5.G, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI. NBO7 Website: https://nbo7.chem.wisc.edu/ or https://nbo6.chem.wisc.edu/nboman.pdf
Reed AE, Weinhold F (1985) Natural localized molecular orbitals. J Chem Phys 83(4):1736–1740. https://doi.org/10.1063/1.449360
Chang TC, Chin J (1996) The Bonding Nature of the Canonical Molecular Orbitals in Simple Diatomic Molecules. Chem Soc TAIP 43(1):1–5
Weinhold F, Landis CR (2001) Natural Bond Orbitals and Extensions of Localized Bonding Concepts. J Chem Educ Res Pract Eur 2:91–104
Weinhold F, Landis CR (2012) (2012) Discovering Chemistry with Natural Bond Orbitals. Wiley, New York
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Landis CR, Weinhold F (2013) NBO 6.0. University of Wisconsin, Boston Theoretical Chemistry Institute, Madison, WI
Ilkhani AR (2020) Non-planarity in four-membered homo-cyclic compounds A4 (A = O, S, Se, Te, Po) and restoring their planarity: a study of the pseudo-Jahn–Teller effect. Theor Chem Acc 139:99
Mahmoudzadeh G, Ghiasi R, Pasdar H (2019) Computational Investigation of the Pseudo Jahn-Teller Effect on the Structure and Chemical Properties of PERHALOETHENE Anions. J Struct Chem 60(5):736–745
Bersuker IB (2021) The Jahn-Teller and Pseudo-Jahn–Teller Effects: AUnique and Only Source of Spontaneous Symmetry Breaking in Atomic Matter. Symmetry 13:1577
Ilkhani AR (2019) Manipulation of planar structure of 1,2,5- and 1,3,4-triazoles and the pseudo Jahn-Teller effect in their 1-pnictogen derivatives. Chem Pap 73:85–94
Gorinchoy NN (2018) Buckybowl Structure of Sumanenes and Distortions of Thiophenes Induced by the Pseudo Jahn-Teller Effect. J Phys Conf Ser 1148:012005
Gorinchoy NN, Ogurtsov IY, Arsene I (2008) Vibronic Origin of the “SKEWED” anticline Configuration of the Hydrogen Peroxide Molecule. Chem J Mold 3(1):105–111
Bersuker IB (2013) Pseudo-Jahn–Teller Effect—A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem Rev 113:1351–1390
Bersuker IB (2021) Jahn−Teller and Pseudo-Jahn−Teller Effects: From Particular Features to General Tools in Exploring Molecular and Solid State Properties. Chem Rev 121:1463–1512
Ilkhani AR (2019) The symmetry breaking phenomenon in heteronine analogues due to the pseudo Jahn-Teller effect. J Mol Model 25:8
Ilkhani AR, Hermoso W (2020) Manifestation of descent symmetry phenomena in tetrahedral structure of M42+ (M = P, As, Sb) analogues. Bull Mater Sci 43:293
Bersuker IB (2012) Pseudo Jahn-Teller Origin of Perovskite Multiferroics, Magnetic-Ferroelectric Crossover, and Magnetoelectric Effects: The d0–d10 Problem. Phys Rev Lett 108:137202
Longuet-Higgins HC, Opik U, Price MHL, Sack RA (1958) Studies of the Jahn-Teller effect. II. The dynamical problem. Proc R Soc London A 244:1–16
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88(6):899–926
Carter-Fenk K, Mundy CJ, Herbert JM (2021) Natural Charge-Transfer Analysis: Eliminating Spurious Charge-Transfer States in Time-Dependent Density Functional Theory via Diabatization, with Application to Projection-Based Embedding. J Chem Theory Comput 17:4195–4210
Ghanbarpour P, NoriShargh D (2016) Exploring the origin of the anomeric relationships in 2-cyanooxane, 2-cyanothiane, 2-cyanoselenane and their corresponding isocyano isomers. Correlations between hyper-conjugative anomeric effect, hardness and electrostatic interactions. RSC Adv 6:46406–46420
Chattaraj PK (1996) The Maximum hardness principle: An overview. Proc Indian Natl Sci Acad 62A(6):513–531
Glossman-Mitnik D (2013) Computational study of the chemical reactivity properties of the Rhodamine B molecule. Procedia Computer Science 18:816–825
Chattaraj PK, Chakraborty A, Giri S (2009) Net electrophilicity. J Phys Chem A 113(37):10068–10074
Rezvani M, Ganji MD, Jameh-Bozorghi S, Hиaзи A (2018) DFT/TD- semiempirical study on the structural and electronic properties and absorption spectra of supramolecular fullerene-porphyrine-metalloporphyrine triads based dye-sensitized solar cells. Spectrochim Acta A Mol Biomol Spectrosc 194:57–66
Sarazin Y Chapple P M. (2022b) Calcium, strontium and barium complexes in organic synthesis. In Elsevier eBooks (pp. 104–192). https://doi.org/10.1016/B978-0-12-820206-7.00069-X
Ghahramanpour M, Jamehbozorgi S, Rezvani M (2020) The effect of encapsulation of lithium atom on supramolecular triad complexes performance in solar cell by using theoretical approach. Adsorption 26(3):471–489
Chattaraj PK, Sarkar U, Roy DR (2006) Electrophilicity Index. Chem Rev 106(6):2065–2091
Jameh-Bozorghi S, Ghahramanpour M, Rezvani M (2022) The role of insertion of Li atom in C60-Porphyrin-Metalloporphyrin, M = (V, Cr, Ni, Cu) as dyes in the DSSC by using the theoretical outlook. Int J New Chem 2:102–128
Komorowski L (1987) Electronegativity and hardness in the chemical approximation 114(1):55–71
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105(26):7512–7516
Pearson RG, Palke WE (1992) Support for a principle of maximum hardness. J Phys Chem 96(8):3283–3285
Pearson RG (1987) Recent advances in the concept of hard and soft acids and bases. J Chem Educ 64(7):561
Parr RG, Donnelly RA, Levy M, Palke WE (1978) Electronegativity: The Density Functional Viewpoint. J Chem Phys 68:3801–3807
Flores-Holguı́N N, Salas-Leiva J, Glossman-Mitnik D. (2023) Computational Discovery of Marine Molecules of the Cyclopeptide Family with Therapeutic Potential. Pharm 16(10):1377
Moghim MT, Jamehbozorgi S, Rezvani M, Ramezani M (2022) Computational investigation on the geometry and electronic structures and absorption spectra of metal-porphyrin-oligo- phenyleneethynylenes-[60] fullerene triads. Spectrochim Acta A Mol Biomol Spectrosc 280:121488
Martínez-Araya JI, Salgado-Morán G, Glossman-Mitnik D (2013) Computational Nanochemistry Report on the OxiCAMs—Conceptual DFT indices and chemical reactivity. J Phys Chem B 117(21):6339–6351
Miller JS, Calabrese JC, Rommelmann H, Chittipeddi SR, Zhang JH, Reiff WM, Epstein AJ (1987) Ferromagnetic behavior of [Fe(C5Me5)2]+ .bul. [TCNE]- .bul. Structural and magnetic characterization of decamethylferrocenium tetracyanoethenide, [Fe(C5Me5)2]+ .bul. [TCNE]- .bul.. cntdot.MeCN and decamethylferrocenium pentacyanopropenide,[Fe(C5Me5)2]+ .bul. [C3(CN)5]. J Am Chem Soc 109(3):769–781
Acknowledgements
The authors thank Professor Dr Davood Nori-Shargh for very useful and instructive guidance.
Funding
No Funding.
Author information
Authors and Affiliations
Contributions
Ghazaleh Kouchakzadeh and Golrokh Mahmoudzadeh carried out computational calculations and analyzed them together, and finalized the results of the manuscript. Ghazaleh Kouchakzadeh wrote the manuscript. The authors were contributed in this research work.
Corresponding author
Ethics declarations
Ethical approval
Ethical approval is not applicable for this research work.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kouchakzadeh, G., Mahmoudzadeh, G. The Pseudo Jahn–Teller effect and NBO analysis for untangling the symmetry breaking in the planar configurations of M2X4+ (M = Si, Ge and X = Cl, Br, I): effect on electronic structure and chemical properties. J Mol Model 30, 1 (2024). https://doi.org/10.1007/s00894-023-05792-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05792-1