
Abstract
Context
The bandgap of metal monochalcogenides (MMCs) is a key property that governs their physical and chemical properties. Accurate measurement of the bandgap is essential for a range of applications, including optoelectronics and photovoltaics. However, many theoretical approximations fail to accurately calculate the bandgap for MMCs, making it difficult to obtain precise values. This study investigated the suitability of the FP-LAPW/GAM-MPW1K scheme for determining the bandgap of MMCs. The investigation included lattice parameters, bandgap, band structure, and density of states, which were compared against both previous theoretical calculations and available experimental data. The findings of the study indicate that the FP-LAPW/GAM-MPW1K approach accurately calculates the bandgap value of MMCs by efficiently treating d-state electrons. The results are consistent with prior studies, confirming the method's reliability in determining the bandgap of these semiconductors.
Methods
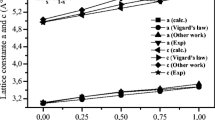
our study used the GAM-MPW1K functional and the full potential linearized augmented plane wave method (FP-LAPW) in the ELK code to calculate the lattice parameters, electronic band structure, and bandgap of ZnS, ZnSe, ZnTe, CdS, CdSe, and CdTe compounds in the wurtzite structure. The crystallographic data were obtained from the COD database and the inputs were prepared by CIF2CELL code. The results were visualized using Xmgrace and VESTA software.





Similar content being viewed by others

Data availability
The data could be accessed from the authors upon request.
References
Brédas JL (1994) Molecular geometry and nonlinear optics. Science 263:487–488. https://doi.org/10.1126/science.263.5146.487
Erbarut E (2003) Optical response functions of ZnS, ZnSe, ZnTe by the LOM method. Solid State Commun 127:515–519. https://doi.org/10.1016/S0038-1098(03)00349-1
Yu Y, Zhou J, Han H et al (2009) Ab initio study of structural, dielectric, and dynamical properties of zinc-blende ZnX (X=O, S, Se, Te). J Alloy Compd 471:492–497. https://doi.org/10.1016/j.jallcom.2008.04.039
Wei S, Lu J, Qian Y (2008) Density functional study of 2D semiconductor CdSe· hda0.5 ( hda = 1,6-hexanediamine) and Its excitonic optical properties. Chem Mater 20:7220–7227. https://doi.org/10.1021/cm703406c
and, and, (2014) First principle studies of structural, elastic, electronic and optical properties of Zn-chalcogenides under pressure. J Semicond 35:072001. https://doi.org/10.1088/1674-4926/35/7/072001
Sörgel J, Scherz U (1998) Ab initio calculation of elastic constants and electronic properties of ZnSe and ZnTe under uniaxial strain. Eur Phys J B 5:45–52. https://doi.org/10.1007/s100510050417
Huynh WU, Dittmer JJ, Alivisatos AP (2002) Hybrid nanorod-polymer solar cells. Science 295:2425–2427. https://doi.org/10.1126/science.1069156
Safari M, Izadi Z, Jalilian J et al (2017) Metal mono-chalcogenides ZnX and CdX (X=S, Se and Te) monolayers: chemical bond and optical interband transitions by first principles calculations. Phys Lett A 381:663–670. https://doi.org/10.1016/j.physleta.2016.11.040
Aras M, Kılıç Ç (2014) Combined hybrid functional and DFT+ U calculations for metal chalcogenides. J Chem Phys 141:044106. https://doi.org/10.1063/1.4890458
Wan Z, Wang Q-D, Liu D, Liang J (2021) Effectively improving the accuracy of PBE functional in calculating the solid band gap via machine learning. Comput Mater Sci 198:110699. https://doi.org/10.1016/j.commatsci.2021.110699
Nourbakhsh Z (2010) Structural, electronic and optical properties of ZnX and CdX compounds (X=Se, Te and S) under hydrostatic pressure. J Alloy Compd 505:698–711. https://doi.org/10.1016/j.jallcom.2010.06.120
Saha S, Pal S, Sarkar P et al (2012) A complete set of self-consistent charge density-functional tight-binding parametrization of zinc chalcogenides (ZnX; X=O, S, Se, and Te). J Comput Chem 33:1165–1178. https://doi.org/10.1002/jcc.22945
Khenata R, Bouhemadou A, Sahnoun M et al (2006) Elastic, electronic and optical properties of ZnS, ZnSe and ZnTe under pressure. Comput Mater Sci 38:29–38. https://doi.org/10.1016/j.commatsci.2006.01.013
Khan I, Ahmad I, Aliabad HAR, Maqbool M (2015) DFT-mBJ studies of the band structures of the II-VI semiconductors. Mater Today: Proc 2:5122–5127. https://doi.org/10.1016/j.matpr.2015.11.008
Singh DJ (1994) Planewaves, Pseudopotentials and the LAPW Method. Springer US, Boston
Yu HS, Zhang W, Verma P et al (2015) Nonseparable exchange–correlation functional for molecules, including homogeneous catalysis involving transition metals. Phys Chem Chem Phys 17:12146–12160. https://doi.org/10.1039/C5CP01425E
Lynch BJ, Fast PL, Harris M, Truhlar DG (2000) Adiabatic Connection for Kinetics. J Phys Chem A 104:4811–4815. https://doi.org/10.1021/jp000497z
Source Forge (2023) The Elk Code. https://sourceforge.net/projects/elk/files/
Lehtola S, Steigemann C, Oliveira MJT, Marques MAL (2018) Recent developments in libxc — A comprehensive library of functionals for density functional theory. SoftwareX 7:1–5. https://doi.org/10.1016/j.softx.2017.11.002
Merkys A, Vaitkus A, Grybauskas A et al (2023) Graph isomorphism-based algorithm for cross-checking chemical and crystallographic descriptions. J Cheminform 15:25. https://doi.org/10.1186/s13321-023-00692-1
Vaitkus A, Merkys A, Gražulis S (2021) Validation of the crystallography open database using the crystallographic information framework. J Appl Crystallogr 54:661–672. https://doi.org/10.1107/S1600576720016532
Quirós M, Gražulis S, Girdzijauskaitė S et al (2018) Using SMILES strings for the description of chemical connectivity in the Crystallography Open Database. J Cheminform 10:23. https://doi.org/10.1186/s13321-018-0279-6
Merkys A, Vaitkus A, Butkus J et al (2016) COD::CIF::Parser : an error-correcting CIF parser for the Perl language. J Appl Crystallogr 49:292–301. https://doi.org/10.1107/S1600576715022396
Gražulis S, Merkys A, Vaitkus A, Okulič-Kazarinas M (2015) Computing stoichiometric molecular composition from crystal structures. J Appl Crystallogr 48:85–91. https://doi.org/10.1107/S1600576714025904
Gražulis S, Daškevič A, Merkys A et al (2012) Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res 40:D420–D427. https://doi.org/10.1093/nar/gkr900
Gražulis S, Chateigner D, Downs RT et al (2009) Crystallography Open Database – an open-access collection of crystal structures. J Appl Crystallogr 42:726–729. https://doi.org/10.1107/S0021889809016690
Downs RT, Hall-Wallace M (2003) The American Mineralogist crystal structure database. Am Miner 88:247–250
Björkman T (2011) CIF2Cell: Generating geometries for electronic structure programs. Comput Phys Commun 182:1183–1186. https://doi.org/10.1016/j.cpc.2011.01.013
Turner PJ (2005) XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR 2. https://plasma-gate.weizmann.ac.il/pub/grace/
Momma K, Izumi F (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Cryst 44:1272–1276. https://doi.org/10.1107/S0021889811038970
Bimberg D (1982) Numerical data and functional relationships in science and technology. Teilbd. a: Gruppe 3: Kristall- und Festkörperphysik = Group 3: @Crystal and solid state physics Bd. 17. Halbleiter Physik der Elemente der IV. Gruppe und der III - V Verbindungen / D. Bimberg. Springer, Berlin Heidelberg
Yadav SK, Sadowski T, Ramprasad R (2010) Density functional theory study of Zn X ( X = O, S, Se, Te ) under uniaxial strain. Phys Rev B 81:144120. https://doi.org/10.1103/PhysRevB.81.144120
Yu PY, Cardona M (2010) Fundamentals of Semiconductors: Physics and Materials Properties, 4th ed. 2010 edition. Springer, Berlin; New York
Hashir P, Pradyumnan PP, Wani AF, Kaur K (2022) Experimental and first-principles thermoelectric studies of bulk ZnO. IOP Conf Ser: Mater Sci Eng 1263:012025. https://doi.org/10.1088/1757-899X/1263/1/012025
Adachi S, Taguchi T (1991) Optical properties of ZnSe. Phys Rev B 43:9569–9577. https://doi.org/10.1103/PhysRevB.43.9569
Christensen NE, Christensen OB (1986) Electronic structure of ZnTe and CdTe under pressure. Phys Rev B 33:4739–4746. https://doi.org/10.1103/PhysRevB.33.4739
Ghahramani E, Moss DJ, Sipe JE (1991) Full-band-structure calculation of first-, second-, and third-harmonic optical response coefficients of ZnSe, ZnTe, and CdTe. Phys Rev B 43:9700–9710. https://doi.org/10.1103/PhysRevB.43.9700
Sharma S, Verma AS, Sarkar BK et al (2011) First principles study on the elastic and electronic properties of CdX (X = S, Se and Te). pp 229–230. https://doi.org/10.1063/1.3653693
Lee G-D, Lee MH, Ihm J (1995) Role of d electrons in the zinc-blende semiconductors ZnS, ZnSe, and ZnTe. Phys Rev B 52:1459–1462. https://doi.org/10.1103/PhysRevB.52.1459
Li ZQ, Pötz W (1992) Electronic density of states of semiconductor alloys from lattice-mismatched isovalent binary constituents. Phys Rev B 46:2109–2118. https://doi.org/10.1103/PhysRevB.46.2109
Yuriychuk I, Fochuk P, Bolotnikov A, James RB (2019) Ab initio GGA+U investigations of the structural, electronic, and magnetic properties of Cd1-xMnxTe alloy. In: Burger A, James RB, Payne SA (eds) Hard X-Ray, Gamma-Ray, and Neutron Detector Physics XXI. SPIE, San Diego, p 65
Wu Y, Chen G, Zhu Y et al (2015) LDA+U/GGA+U calculations of structural and electronic properties of CdTe: dependence on the effective U parameter. Comput Mater Sci 98:18–23. https://doi.org/10.1016/j.commatsci.2014.10.051
Hussain MI, Khalil RMA, Hussain F (2021) Computational exploration of structural, electronic, and optical properties of novel combinations of inorganic Ruddlesden-popper layered perovskites Bi 2 XO 4 (X = Be, Mg) using Tran and Blaha-Modified Becke-Johnson approach for optoelectronic applications. Energy Tech 9:2001026. https://doi.org/10.1002/ente.202001026
Hussain MI, Khalil RMA, Hussain F, Rana AM (2021) DFT -based insight into the magnetic and thermoelectric characteristics of XTaO 3 (X = Rb, Fr) ternary perovskite oxides for optoelectronic applications. Int J Energy Res 45:2753–2765. https://doi.org/10.1002/er.5968
Hussain MI, Khalil RMA, Hussain F et al (2020) Investigations of structural, electronic and optical properties of TM-GaO 3 (TM = Sc, Ti, Ag) perovskite oxides for optoelectronic applications: a first principles study. Mater Res Express 7:015906. https://doi.org/10.1088/2053-1591/ab619c
Acknowledgements
The authors confirm that the study was conducted in an ethical and responsible manner. A part of this work was granted access to the HPC ressources of UCI-UFMC (Unité de Calcul intensif of the University FRERES MENTOURI CONSTANTINE1).
Funding
The authors, A. T. and A. M., are affiliated with Constantine 1-Freres Mentouri University, Algeria. The authors declare that this research was conducted for academic purposes and was not influenced by any external funding or organization.
Author information
Authors and Affiliations
Contributions
A. T and A. M. contribute equally in this paper.
Corresponding author
Ethics declarations
Ethical approval
The authors declare that they followed all ethical guidelines while conducting the research and preparing the manuscript. The authors declare that the research work presented in this paper is original.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Toumiat, A., May, A. The FP-LAPW/GAM-MPW1K approach: a reliable abinitio method for calculating the band gap of II-VI semiconductors monochalcogenides. J Mol Model 29, 297 (2023). https://doi.org/10.1007/s00894-023-05696-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05696-0