
Abstract
[FeFe]-hydrogenases are gas-processing metalloenzymes that catalyze H2 oxidation and proton reduction (H2 release) in microorganisms. Their high turnover frequencies and lack of electrical overpotential in the hydrogen conversion reaction has inspired generations of biologists, chemists, and physicists to explore the inner workings of [FeFe]-hydrogenase. Here, we revisit 25 years of scientific literature on [FeFe]-hydrogenase and propose a personal account on ‘must-read’ research papers and review article that will allow interested scientists to follow the recent discussions on catalytic mechanism, O2 sensitivity, and the in vivo synthesis of the active site cofactor with its biologically uncommon ligands carbon monoxide and cyanide. Focused on—but not restricted to—structural biology and molecular biophysics, we highlight future directions that may inspire young investigators to pursue a career in the exciting and competitive field of [FeFe]-hydrogenase research.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen turnover (\({\mathrm{H}}_{2}\rightleftharpoons 2 {\mathrm{H}}^{+}+2 {\mathrm{e}}^{-}\)) is an important reaction in microorganisms that thrive under reducing, anoxic conditions and plays a role in certain aerobes [1]. Trace amounts of H2 provide electrons to power the anabolism of numerous archaea and bacteria in the soil, aqueous environments, or host tissue [2,3,4]. Under fermentative or ‘micro aerobic’ conditions, proton reduction and H2 release has been shown to be a key in the redox regulation of autotrophs like photosynthetic bacteria and algae [5,6,7]. The enzymes responsible for proton reduction and H2 oxidation are referred to as ‘hydrogenases’ [8,9,10].
In archaea, Hmd [Fe]-hydrogenase is involved in the conversion of CO2 to CH4 (methanogenesis), catalyzing H2 splitting and hydride transfer at a cofactor with a monometallic iron center [11,12,13]. Unlike Hmd [Fe]-hydrogenase, [NiFe]- and [FeFe]-hydrogenase are iron-sulfur enzymes and carry a bimetallic active site cofactor. [NiFe]-hydrogenases have been found in archaea and bacteria, and are typically involved in heterotrophic H2 uptake [13,14,15]. The soluble ‘standard’ [NiFe]-hydrogenases are distinguished from membrane-bound, multimeric, as well as O2-tolerant and bidirectional [NiFe]-hydrogenases [15,16,17]. Although highly diverse, all [NiFe]-hydrogenases share the same active site cofactor. It is comprised of a nickel and an iron ion, covalently attached to the enzyme by either four cysteine residues, or three cysteine and one selenocysteine. The iron ion is coordinated by two cyanide (CN–) and one carbon monoxide ligand (CO) ligand, the latter being a common feature of hydrogenases [18].
[FeFe]-hydrogenases have been found in bacteria, lower eukaryotes, and green algae, representing the phylogenetically most recent class [8,9,10]. [FeFe]-hydrogenases are involved in H2 uptake as well as redox regulation (H2 release upon proton reduction), but in complex with other metalloproteins, additional roles in CO2 reduction or electron bifurcation are suggested [19]. Figure 1 depicts cyclic voltammograms of two different [FeFe]-hydrogenases that demonstrate bidirectional catalysis [20]. Typically, H2 oxidation is observed up until 0 V vs SHE, while many [FeFe]-hydrogenase are inhibited at more positive potentials, i.e., due to an interaction with halide anions like chloride [21]. At potentials more negative than -0.3 V, a proton reduction current is observed resulting in H2 evolution while the current at higher potentials indicates H2 oxidation. Near the equilibrium or midpoint potential (Em) the ‘clean cut’ through the zero-current X-axis indicates minimum electric overpotential, a key property of hydrogenase-catalyzed hydrogen turnover [22,23,24]. The catalytic mechanism of [FeFe]-hydrogenase is discussed controversially [25,26,27] as we will see in Sect. “The Spectroscopic Characterization of [FeFe]-hydrogenase”.

Reproduced with permission from the ACS
Cyclic voltammograms of [FeFe]-hydrogenases A CrHydA1 and B CaHydA indicate bidirectional catalysis. Recorded between −0.5 V and +0.3 V vs. SHE at pH 6 and in the presence of 1 atm H2, (i) positive currents and (ii) negative currents results from H2 oxidation and proton reduction (i.e., H2 evolution), respectively. The midpoint potentials (Em) are highlighted. The decrease of current above 0 V (particularly visible for CrHydA1 in panel A) is due to ‘anaerobic inactivation’. [20]
While all [FeFe]-hydrogenases carry the same catalytic cofactor, a classification into four groups A–D has been suggested among which ‘Group A’ represents the well-studied ‘standard’ [FeFe]-hydrogenases [28]. Figure 2 shows the crystal structure of standard [FeFe]-hydrogenase CpI including the four ferredoxin-type iron-sulfur clusters (‘F-clusters’) that connect the hydrogen-forming active site cofactor (‘H-cluster’) with the surface. The H-cluster comprises a [4Fe-4S] cluster (‘[4Fe]H’), connected to a homobimetalic iron site via a bridging cysteine ligand. The metal ions of this diiron site (Fep, Fed) are modified with a bridging carbonyl ligand (µCO) and two terminal CN– and CO ligands, while the apical-binding site of Fed (X) may be vacant or occupied with different ligands. A unique S2(CH2)NH ligand (azadithiolate, ADT) connects the two iron ions. The biosynthesis of the H-cluster is well understood [29].

Crystal structure of the [FeFe]-hydrogenase CpI (PDB ID 4XDC). The accessory, ferredoxin-type iron-sulfur clusters (F-clusters) are highlighted in blue, the active site cofactor (H-cluster) is highlighted in red. On the right side, the H-cluster is shown as a chemical model. Position ‘X’ may be vacant or occupied with ligands like CO, CN–, H–, S–, or O2–. [4Fe]H, [4Fe-4S] cluster; µCys, bridging cysteine; CO, carbon monoxide; CN–, cyanide; ADT, azadithiolate; Fep/Fed proximal/distal iron ion (relative to [4Fe]H)
The active site niche of the H-cluster provides a specialized environment beyond the first coordination sphere [30]. The [4Fe]H cluster is in electron tunneling distance to the proximal [4Fe-4S] cluster, facilitating electron exchange between the diiron site and soluble redox partners like ferredoxin [31] The charge of the F-clusters affects the electron density distribution across the H-cluster and has been suggested to influence catalysis [32], together with protein structural fluctuations [33] and/or protonation changes near the [4Fe]H. [34] Conserved amino acid residues facilitate proton transfer between bulk solvent and H-cluster [35] Here, the amine head group of the ADT ligand receives a proton from a nearby cysteine (Fig. 2). The ADT ligand does not only function as a proton relay but has been discussed to stabilize various H-cluster states by ‘intramolecular’ hydrogen bonding [36] The CO and CN– ligands have be modeled to face hydrophobic and hydrophilic pockets of the active site niche, and for Fep-CN–, hydrogen-bonding interactions with the protein fold have been confirmed [37] While the H-cluster is the same in all [FeFe]-hydrogenases, significant diversity is observed among the primary structure of Group A–D enzymes [28].
In this review, we will establish a historical and topical overview on the literature of [FeFe]-hydrogenase since the late 1990s. We focus on structural biology and biophysical aspects of hydrogenase research. The timeline in Scheme 1 suggests four phases: between 1998 and 2002 (phase I), the first crystal structures were solved by X-ray diffraction protein crystallography and three H-cluster states (Hox, Hox-CO, Hred) were characterized by electron paramagnetic resonance spectroscopy and Fourier-transform infrared spectroscopy. In a second phase (ca. 2004–2010), different bacterial expression hosts were introduced that allowed producing and modifying [FeFe]-hydrogenases in preparative amounts. Moreover, protein film electrochemistry became a go-to technique following hydrogenase turnover, CO inhibition, and O2 deactivation. The understanding of in vivo H-cluster synthesis and the development of ‘artificial maturation’ characterize the third phase (ca. 2010–2017). The characterization of cofactor states with a reduced [4Fe]H cluster (Hsred, Hred´) falls in this phase, too. The current and ongoing phase IV (since 2017) is dominated by the discussion about proton transfer, electron transfer, and the catalytic mechanism of [FeFe]-hydrogenase. Additionally, we saw a ‘revival’ of the inactive H-cluster state (Hinact) that protects [FeFe]-hydrogenase from O2 deactivation.

Timeline of important events and key findings in [FeFe]-hydrogenase research. Phases I–IV are marked black, blue, red, and white, respectively. Hox, Hox-CO, Hred, Hsred, Hred´, and Hinact are H-cluster states
This review is organized in two main sections, introducing structural aspects of [FeFe]-hydrogenase in Sect. “The Structural Characterization of [FeFe]-hydrogenase” and spectroscopic properties in Sect. “The Spectroscopic Characterization of [FeFe]-hydrogenase”. Following each section or sub-section, ‘further reading’ suggestions will serve as starting point for a topical deep dive into the literature on [FeFe]-hydrogenase.

The structural characterization of [FeFe]-hydrogenase
We will start this chapter with a brief summary of key findings between 1931 and 1997; for a more detailed review on the history of [FeFe]-hydrogenase until 1990, see Ref. [38]. Hydrogenases were first described by Stephenson and Stickland in 1931 [39] and demonstrated to catalyze H2 oxidation and H/D exchange with the solvent by Yudkin et al. in 1934 [40]. Throughout the 1940’s and 1950’s, hydrogenases were reported to interact with O2, CO, CN–, and visible light [41] and although the prosthetic groups were unknown, non-heme iron was identified to play a key role [42]. In 1963, Sadana and Rittberg noted a peculiar dependence of hydrogenase activity on sulfur, which led them to postulate ferrous-hydroxyl complexes [43]. Improving the purity of enzyme preparations in the early 1970’s, Nakos and Mortenson eventually reported an iron-sulfur ratio of 1:1 in the hydrogenase from C. pasteurianum [44], whose electron paramagnetic resonance (EPR) spectra resembled the [4Fe-4S] clusters of ferredoxin [45] While this settled the identity of hydrogenases as iron-sulfur enzymes, EPR also suggested the presence of a unique metal site, tentatively assigned to the catalytic active site in an important review article by Mortenson et al. [46]. In the 1980’s, certain hydrogenase preparations showed evidence of [3Fe-4S] clusters [47], selenocysteine [48], and a dependence on nickel [49], which inspired the distinction between “Fe-hydrogenase” and “Ni-hydrogenase” (Hmd [Fe]-hydrogenase was identified by Thauer et al. no earlier than 1992 [50]).
The first structure of a [NiFe]-hydrogenase was published by Fontecilla-Camps in 1995, showing the active site iron ion with three non-protein ligands, tentatively assigned to water [51] Based on the unique infrared signature of [NiFe]-hydrogenase, in 1997, Albracht et al. suggested these ligands to be CO and CN– [52], and comparing the infrared spectra of different hydrogenases, a somewhat similar architecture was predicted for [FeFe]-hydrogenase [53], the later which was yet to be crystalized.
In December 1998 and January 1999, the first cryogenic X-ray diffraction (XRD) crystal structures of [FeFe]-hydrogenase were published independently by two groups. Peters et al. solved the structure of the oxidized, monomeric [FeFe]-hydrogenase from C. pasteurianum (CpI) with a resolution of 1.8 Å [54] The authors identified the binding motives of the accessory iron-sulfur F-clusters that facilitate electron transfer between bulk solvent and active site cofactor. Furthermore, Peters et al. were the first to describe atomistic details of the H-cluster, including a discussion of amino acid residues that line the active site niche and may serve as hydrogen-bonding partner to the CN– ligands, shield the H-cluster from unwanted solvent access, or enable catalytic proton transfer. It took 20 more years to validate the functionality of the proton transfer pathway in [FeFe]-hydrogenase CpI [55,56,57]. Here, protein crystallography helped uncovering the role of water molecules that substitute for certain amino acid sidechains when replaced by site-directed mutagenesis. In their pioneering study, Peters et al. found the H-cluster to bind a putative water molecule in the apical position of the distal iron ion, Fed (Fig. 3A). Fontecilla-Camps et al. published the crystal structure of the oxidized, dimeric [FeFe]-hydrogenase from D. desulfuricans (DdHydAB) with a resolution of 1.6 Å only one month later [58] Their model of the H-cluster shows no indications of a water molecule in the apical position of Fed. Furthermore, the structure comprises a fully resolved dithiolate ligand, which was assigned to S2(CH2)3 back then (propanedithiolate, PDT, see Fig. 3B). Fontecilla-Camps et al. observed a hydrophobic gas channel and discussed an alternative proton transfer pathway, notably independent of the dithiolate ligand and including a conserved lysine residue in hydrogen-bonding distance to the distal CN– ligand. Although certain conclusions from this study did not stand the test of time, the closing statement reads virtually prophetic:
“Perhaps the most striking feature of the novel diiron active-site center of the [FeFe]- hydrogenase is its almost complete independence from protein coordination. It is possible that such a center may have been imported from the inorganic world as an already functional unit.”

Early XRD crystal structures of the H-cluster of [FeFe]-hydrogenases CpI and DdHydAB in the oxidized (A, B), CO-inhibited (C), and reduced state (D). The identity of the dithiolate ligand, PDT, was latter corrected to ADT. From A–C, the PDB IDs are 1FEH, 1HFE, and 1C4A (unfortunately, there is no PDB entry for reduced DdHydAB). Reproduced with permission from the AAAS [54], Cell Press [58], and the ACS [59, 60]
In the following years, the group of John Peters published on the mechanism of CO inhibition of [FeFe]-hydrogenase. Studying the photochemistry of CO-inhibited CpI in cristallo [59] established the belief that external CO binds at the apical-binding site of Fed (Fig. 3C). Meanwhile, the group of Juan Fontecilla-Camps investigated the geometry of the H-cluster of DdHydAB crystals grown in the presence of chemical reductant and H2 [60]. Their XRD model of the reduced H-cluster comprised an open apical coordination site and a ‘semi-bridging’ CO ligand at Fed (Fig. 3D). The latter is in variance to the oxidized H-cluster that clearly showed a Fe–Fe bridging CO ligand, µCO [54, 58]. The crystallographic findings were in apparent agreement with room temperature infrared spectro-electrochemistry titrations on [FeFe]-hydrogenase solution that showed the loss of µCO upon conversion of the oxidized state Hox into one of the reduced states, Hred [60]. Accordingly, Fontecilla-Camps et al. assigned the ‘semi-bridging’ H-cluster geometry to Hred, a decision that would dominate the interpretation of [FeFe]-hydrogenase catalysis for nearly 20 years.
The rise of the photosynthetic hydrogenase and artificial maturation
Across the first decade of the twenty-first century, the enzyme CrHydA1 from green algae Chlamydomonas reinhardtii became the new standard [FeFe]-hydrogenase. Happe and Stripp chose the catchphrase “photosynthetic hydrogenases”, because it accepts electrons from ferredoxin at the reducing end of the photosynthetic electron transport chain [61] CrHydA1 is attractive for many spectroscopic approaches (Sect. “The Spectroscopic Characterization of [FeFe]-hydrogenase”) since it lacks the accessory iron-sulfur clusters of CpI and DdHydAB. Following protocols for homologous and heterologous production in Clostridium acetobutylicum or Shewanella oneidenis [62,63,64], eventually transgenic E. coli cells (carrying the genes of [FeFe]-hydrogenase maturases HypEFG) were established as most efficient expression host for CrHydA1 [65, 66]. In 2010, Peters et al. exploited the E. coli protocol to produce and crystallize apo-CrHydA1, i.e., enzyme without the diiron site of the H-cluster [67]. Despite various efforts, the structure of functional holo-CrHydA1 is yet to obtain; it has been argued that the 45 amino acid insertion forms a flexible loop that might be important for the interaction with in vivo redox partner ferredoxin [68]—but ultimately impedes crystallization.
Understanding the enzymatic details of H-cluster biosynthesis was basis for the production of [FeFe]-hydrogenase in E. coli, an organism that naturally relies on [NiFe]-hydrogenase. After the successful modification of E. coli with [FeFe]-hydrogenase maturases HydEFG and the establishment of efficient expression, synthesis, and purification protocols [65,66,67], the next step was to establish the artificial maturation of [FeFe]-hydrogenase [69,70,71]. Fontecave et al. reported that HydF can be loaded with synthetic precursors of the diiron site [69]. When HydF and apo-CrHydA1 was mixed, functional [FeFe]-hydrogenase was produced, which allowed generating ‘cofactor variants’, e.g., using unnatural diiron site precursors (Fig. 4). Less than two months later, Happe et al. demonstrated the direct activation of apo-CrHydA1 and apo-CpI, avoiding HydF altogether [70]. This further simplified the production of native and artificial [FeFe]-hydrogenase. The artificial maturation of DdHydAB was reported as well [71]; however, the majority of cofactor variants has been produced in CrHydA1 and CpI, including isotopomers [72,73,74], variations of the ligands [75], the iron ions [76], and the sulfur atoms [77,78,79]. Numerous CpI cofactor variants have been crystallized [78,79,80].

Synthetic diiron complexes can be used to activate the [FeFe]-hydrogenase maturation enzyme HydF (A) or the [FeFe]-hydrogenase apo-protein directly (B). In the initial studies, a variation of the dithiolate headgroup (X) was demonstrated. Reproduced from Ref. [69] with permission from Nature Springer
Deactivation and inhibition in the presence of O2
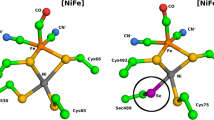
Most [FeFe]-hydrogenases are irreversibly deactivated by trace amounts of O2. Although DdHydAB can be isolated under aerobic conditions [58], once activated the H-cluster falls apart when reacted with O2. This is in marked difference to [NiFe]-hydrogenase that are reversibly inhibited by O2 or oxidize O2 to H2O maintaining hydrogen turnover under aerobic conditions [81, 82]. The deactivation reaction of [FeFe]-hydrogenase is less well understood. Crystal structures of CpI obtained at different time points after exposure to O2 point toward oxidative damage at the H-cluster, possibly due a formation of reactive oxygen species (ROS), as suggested in earlier work [83,84,85]. Catalytic conversion of O2 into ROS is supported by the observation that CO inhibits both the reaction with H2 and O2 [20] hinting at a similar binding site, i.e., the open coordination site of Fed. ‘Super-oxidized’ intermediates of the reaction with O2 are typically short-lived, but proton transfer variants like C169A of CrHydA1 allowed stabilizing respective H-cluster species [86]. This hints at H2O2 as a key ROS in the deactivation process. The structure of the H-cluster in the O2-inhibited state Hinact of DdHydAB and CbA5H was debatted until crystallographic analyses revealed that a cysteine residue binds to Fed of the H-cluster, protecting the cofactor from O2 deactivation like a ‘safety cap’ [87]. Interestingly, sulfide has a similar effect and binds to Fed under oxidizing conditions both aerobic and anaerobic (Fig. 5), which allows reversible conversion of the Hox and Hinact states [88,89,90].

XRD crystal structure of DdHydAB (PDB ID 6SG2) and CbA5H (PDB ID 6TTL) in the Hinact state. A In DdHydAB, the apical-binding site of Fed indicates electron density subsequently identified as a sulfur atom by XAS spectroscopy. Reproduced from Ref. [91] with permission from Wiley–VCH. B In CbA5H, adjacent cysteine C367 binds to the apical binding site of Fed in the presence of O2. Reproduced from Ref. [87] with permission from Springer Nature
Future directions
Despite the pioneering work on CO-inhibited and H2-reduced [FeFe]-hydrogenase no other redox state has been explored by protein crystallography in the last 20 years. Certain updates are necessary. For example, to conclude the discussion regarding the identity of the diiron site-reduced redox states Hred and Hsred (Sect. “The Spectroscopic Characterization of [FeFe]-hydrogenase”), it will be required to obtain atomic resolution XRD data on spectroscopically verified protein crystals, i.e., using in cristallo spectroscopy [92, 93]. The effect of temperature will have to be considered as well [94]; to this end, King and Peters et al. published the first XRD structure of oxidized CpI using the X-ray free electron laser (XFEL) at Linac Coherent Light Source, which allows recording radiation damage-free diffraction patterns [33]. The authors measured CpI at cryogenic temperature [33] but in principle, protein structure determination at ambient-temperature is possible using XFEL radiation. Low temperature cannot be avoided in cryogenic electron microscopy (Cryo-EM) experiments and the technique may not be suitable for atomistic details and the comparatively small standard [FeFe]-hydrogenases DdHydAB, CpI, and CrHydA1. However, [FeFe]-hydrogenases are diverse, and there are multimeric enzymes that are poorly understood [19]. Just recently, the Cryo-EM structures of HydABC from Thermotoga maritima, Acetobacterium woodii, and Thermoanaerobacter kivui have been employed to understand the mechanism of electron bifurcation [95, 96]. To conclude this section, the potential of ambient-temperature neutron scattering in the determination of hydrogen atoms is noted. This technique may help identifying site-selective protonation or hydride formation events. Initial work on [NiFe]-hydrogenase has been published [97] but no [FeFe]-hydrogenase structure is reported yet.

The spectroscopic characterization of [FeFe]-hydrogenase
In the second section, we saw how the structural characterization of CpI and DdHydAB by cryogenic XRD lay the foundation for a principle understanding of [FeFe]-hydrogenase function. To rationalize the details of electron transfer, proton transfer, and the catalytic mechanism, additional methods must be taken into consideration. Such methods, electron paramagnetic resonance spectroscopy (Sect. “Electron Paramagnetic Resonance”) and vibrational spectroscopies (Sect. “Vibrational Spectroscopy”), are complimentary techniques which allow for studying stable intermediates along the catalytic cycle of [FeFe]-hydrogenase.
Electron paramagnetic resonance
Electron paramagnetic resonance (EPR) is a spectroscopic technique to investigate metal centers or organic molecules with unpaired electrons. An advantage of EPR spectroscopy is that the experiment specifically reports on the unpaired electron at the active site. In contrast, nuclear magnetic resonance (NMR) spectroscopy probes all the nuclei in a protein and may have a ‘blind sphere’ around a paramagnetic center (i.e., a metalloprotein active site) whereas the NMR signal is unrecognizably broadened. However, for EPR spectroscopy, there are no protein size limitations and, through the interactions of the electron with the environment (first and second coordination sphere, coupled nuclei, etc.), a unique view of the electronic structure of paramagnetic states along an enzymatic catalytic cycle can be obtained [98]. Both conventional continuous wave (CW) and ‘advanced’ pulse EPR spectroscopy are central in metalloprotein research [99] and have been extensively used in the analysis of [FeFe]- and [NiFe]-hydrogenase [100].
Insights from continuous wave and advanced EPR
The active site cofactor and the accessory iron-sulfur clusters of [FeFe]-hydrogenase give rise to characteristic EPR signals that can be stabilized as intermediates along the catalytic cycle. While the F-clusters are paramagnetic in the reduced state, the H-cluster show EPR activity in the oxidized state [101,102,103], which allows for a clear separation of signals between the active site and electron transfer path.
Investigating the F-clusters to understand electron transfer and magnetic coupling, Peters et al. recorded the EPR spectra of apo-CpI and cofactor variant CpIPDT under increasingly reducing conditions [104]. The authors determined the individual second derivative spectra and midpoint redox potentials (Em) of all metal centers (Fig. 6A). The low potential of the histidine-ligated, distal cluster FS4C (Em ≤ −450 mV) will limit the physiological H2 evolution activity of CpI to higher concentrations of ferredoxin; however, the small differences between FS4B, FS4A, and the H-cluster (ΔEm = 10 mV) suggest unimpeded electron transfer in both directions and suggest a large degree of magnetic coupling [104]. Such effects were argued to influence proton transfer, protonation events at the H-cluster, and catalytic turnover in general [105].

A Extracted second derivative EPR spectra of the H-cluster state Hox in cofactor variant CpIPDT and the F-clusters FS4A–C ([4Fe-4S]) and FS2 ([2Fe-2S]). B Fdx/CpI docking model annotated with the midpoint redox potentials of all hydrogenase metal centers. Reproduced from Ref. [104] with permission from the ACS
Once the EPR spectra of the F-clusters are known, the H-cluster can be analyzed specifically. Early work on aerobically isolated DdHydAB showed that the diamagnetic, O2-inhibited state Hinact is activated under reducing conditions. At about −200 mV vs. SHE one electron jumps at the [4Fe]H cluster, forming the Htrans state that converges irreversibly into the Hox state at about −300 mV vs. SHE. This indicates a reduction of the diiron site upon intramolecular electron transfer [106, 107]. Both Htrans and Hox are paramagnetic and give rise to rhombic EPR spectra (Fig. 7). While Hinact and Htrans were initially not observed in other [FeFe]-hydrogenases, several authors recently demonstrated the formation of Hinact over Hox in the presence of sulfide [89, 90]. When Hox is reacted with CO instead, [FeFe]-hydrogenases adopt the Hox-CO state (Fig. 7) that is characterized by an axial EPR spectrum [103] resulting from strong spin exchange coupling between [4Fe]H cluster and diiron site [108]. This is in agreement with the 57Fe hyperfine couplings observed by Mössbauer spectroscopy [109,110,111]. Today, the redox state of the metal ions in the diiron site is commonly denoted as [Fep(II)-Fed(I)] for Hox (with a more equal distribution of charges in Hox-CO) and [Fep(II)-Fed(II)] for Htrans and a recently discovered, cyanide-inhibited H-cluster state [112, 113]. The paramagnetism of the H-cluster in the latter states results from the reduced [4Fe]H cluster, which also explains the EPR spectrum of the so-called hydride state, Hhyd [114,115,116]. Formally, a ‘super-reduced’ H-cluster species, most of the electron density is located at the apical hydride ligand at Fed, stabilized by a [Fep(II)-Fed(II)]-type diiron site and a reduced [4Fe]H cluster. Figure 7 shows that the rhombic EPR spectra of Hhyd and Htrans are strikingly similar but clearly distinct from Hox. Reduction of [4Fe]H and the diiron site ([Fep(I)-Fed(I)]) produces another ‘super-reduced’ state, Hsred, that shows a broad axial EPR signal. Horch and Zebger et al. suggested that fast spin relaxation across the H-cluster in Hsred can explain these spectral properties [117].

Simulation of EPR spectra at 9.5 GHz (X-band) for key paramagnetic H-cluster states. Characteristic g-tensors are annotated as observed in DdHydAB (Htrans) and CrHydA1 (everything else)
In the previous section, we highlighted the use of CW EPR spectroscopy to characterize the stable intermediates of the active site H-cluster and stepwise reduction techniques to follow the electron pathway of the F-clusters. Continuous wave EPR, gives a snapshot of the electron and the total interaction with the coordination environment. This includes broadening effects, strong nuclei coordination, and spin–orbit coupling of the electron to direct coordination (metal ion; rhombicity). However, finer details of the coordination are unable to be determined due to the linewidths of the inhomogenously broadened spectra that is typical of the [FeFe]-hydrogenase active site [98]. Instead, we use pulse EPR techniques to ‘look within’ the broadened spectrum to characterize direct coordination between the free electron and surrounding nuclei which possess a nuclear magnetic moment (57Fe, 14/15N, 13C, 17O, 33S, 31P, 1/2H, etc.).
In these cases double resonance techniques, such as electron nuclear double resonance (ENDOR) where the nuclei are directly excited by an additional incident radio frequency (< 100 MHz) pulse, or multi-pulse techniques using only microwave pulses, such as electron spin echo envelope modulation (ESEEM) and hyperfine sub-level correlation (HYSCORE) spectroscopies are used, where the EPR signal is modulated by the forbidden transition probabilities of the coupled nuclei [118]. These techniques can be performed at multiple frequencies such as, 9.5 and 35 GHz, but also high frequency (> 94 GHz), and provide the framework to study the coordination chemistry of the active site in detail [118]. By increasing the frequency of the EPR experiment, the anisotropy of the g-tensor is increased further resolving the gxx, gyy, and gzz components. This allows for orientation selection to be performed on a frozen solution sample and the g- and hyperfine-tensors can be estimated.
Although the wild-type [2Fe]H cluster contains 14N ligands in the ADT ligand and the two CN– ligands, it would be beneficial to be able to choose the isotopes in the active site to further gain information on the electron interaction within the diiron site. This was achieved by the ability to express [FeFe]-hydrogenase in its apo form, synthesize a chemical mimic of the [2Fe]H cluster, and for the chemical mimic to be inserted in apo-HydA1 producing a functioning enzyme [71, 72]. Doing so allowed the characterization of enriched isotopes (2H, 13C, 15N, 57Fe) of the [2Fe]H cluster to be studied by EPR pulse spectroscopy to learn about the electronic structure of the active site. Additionally, non-natural [2Fe]H clusters could be synthesized to trap intermediates that are typically transient or elusive [119] and give insights into proton transfer pathways [36, 80].
In this section, we will highlight the use of pulse EPR to characterize the active site in the Hox and Hox-CO state. Then, we focus on recent work on characterizing the maturation process of the [2Fe]H cluster using isotope labeled amino acids in HydG, HydE, and HydF. This section ends with a brief overview on where information gaps exist and how further techniques can be exploited on to address these shortcomings.
Characterization of the oxidized H-cluster and Maturation.
The data in Tables 1, 2 represent the current knowledge on the electronic properties of the [FeFe]-hydrogenase in the Hox and Hox-CO states. However, this table is not exhaustive and further information on both the magnitude and orientation of the g-, hyperfine-, and quadrupole-tensors are still needed. These tables are further complicated by the g-value and hyperfine shifts seen between different hydrogenase species, but still serves as a collection of the many hyperfine experiments that have been performed on [FeFe]-hydrogenase [73, 120,121,122]. Since EPR is a direct measurement of the electronic structure of the state of the enzyme, the g- and hyperfine-tensor shifts indicate a perturbation of the electronic structure by the 1st and 2nd coordination sphere. It is this local environment that plays an important role in fine-tuning the overall reactivity of the enzyme [30].
For the [2Fe]H cluster specifically, differences in the g-values and hyperfine-values using 57Fe between species are hypothesized to be variable coupling to the [4Fe]H cluster. Full DFT models using all six iron ions (as opposed to truncated models of the [2Fe]H cluster in ref. [123]) have put the effective electron spin at on the Fed while DFT calculations of Hox-CO place the majority of the electron spin at the Fep. From experimental data, shown in Table 2 and noted in Ref. [124], although most of the electron spin is located on Fep, hyperfine data of the other ligands suggests substantial distribution over Fed and the CO but not over the CN– ligands. This paints an interesting picture of the interaction of the exogenous ligand COexo, the [2Fe]H cluster, and the drastic electronic re-configuration when CO is released forming the active Hox state.
Recent experiments using time-resolved CW EPR by Berggren et al. allowed monitoring the adoption of the [2Fe]H cluster into an apo-HydA protein [128]. These data suggest an incorporation of the diiron site to the apo-HydA1 via electron transfer from [4Fe]H2+ to [4Fe]H+ and changes in the redox state of the Fed ion forming a CO-inhibited [4Fe]H+-[Fep(II)Fed(I)] configuration. This species is the Hred´-CO intermediate [129] that rapidly oxidizes into an [4Fe]H2+–[Fep(II)Fed(I)] configuration, characteristic for the Hox-CO state. This rate-limiting step increases the energy required to remove the [2Fe]H cluster. The final step is the slow release of COexo to form the catalytically active Hox state.
Fundamental work by Britt et al. has put together the biosynthesis model of the [2Fe]H cluster (Fig. 8), using an array of biophysical techniques [130,131,132,133]. Included in these techniques was the isotope labeling of key amino acids in the biosynthetic pathway. Here, the radical S-Adenosyl-L-methionine (rSAM) enzyme, HydG, was found to cleave tyrosine to generate the CO and CN– ligands as the precursor to the [2Fe]H cluster. Extensive CW and pulse EPR was performed by Britt et al. for characterizing this rSAM mechanism [134]. By removing the dangler iron attached to the rSAM [4Fe-4S] active site, it was possible to pause HydG reactivity and study the tyrosine substrate by separately labeling two hydrogen pairs with 2H, and a carbon pair with 13C. By comparing the EPR experimental data with DFT, the coordination chemistry and mechanism of HydG was proposed (Fig. 8). Prior to the understanding of HydG, the rSAM enzyme, HydE, could be studied by providing a synthetic substrate to HydE and watching the maturation with EPR [135] and the individual steps were found, again, with isotope labeling and following maturation through double resonance techniques [130].

Proposed sequential model for the roles of HydG, HydE, and HydF in the biosynthesis of the H-cluster. This model has been developed by David Britt and others using an array of biophysical techniques (EPR, FTIR, XRD, EXAFS). This figure is obtained from Ref. [131] under the creative commons license from RSC
In the studies of HydE, freeze-quenched methods with a time resolution of 10 s were employed to prepare EPR samples using a synthesized HydG product (complex-B) named ‘syn-B’. Letting the intermediate complex evolve over approximately 10 min reveals an EPR active complex that is shown to retain the two CO and one CN– ligands from HydG biosynthesis. Finally, this EPR active intermediate forms an antiferromagnetically coupled (S = 0, EPR silent) [Fe(I)2S2(CO)4(CN)2] dimer intermediate. Future work is required to provide the biosynthesis of the final [2Fe]H cluster with the required ADT ligand.
Through these finding, this work has also led the way in biosynthesis models of [FeFe]-hydrogenase with only HydF maturase [136]. These findings are important for future work in selective isotope labeling of the [2Fe]H cluster and teasing out further mechanistic studies of HydF and HydE. Although the docking of HydE to HydF is known, the product transfer and final maturation is still unknown and will take a combination of FTIR and EPR studies to find the last steps in the biosynthesis.
Ultimately, as this work matures the understanding of the maturation will further improve synthesis protocols of [2Fe]H cluster for bio-inspired mimics. In a recent review, Broderick et al. summarize the biosynthetic pathway of the [FeFe]-hydrogenase, including the semisynthetic approaches by Britt et al. using advanced EPR methods [137].
Future directions
[FeFe]-hydrogenases are under further investigation using spectroscopic techniques to understand the mechanisms of O2 tolerance. Kasanmascheff et al. reported an interesting radical (R•ox) in the [FeFe]-hydrogenase CbA5H, which is known to be O2-tolerant [87, 138]. When kept under aerobic conditions, the R•ox signal significantly increases while CbA5H remains in the Hinact state. Further studies using 57Fe labeling produced hyperfine couplings in the 25–35 MHz range, suggesting a similar configuration to the Hox-CO state. The origin of R•ox needs further experimental and site-directed mutagenesis studies. For instance, in Hinact of DdHydAB, a sulfur has been shown to be coordinated to Fed [91] and a radical formed from a nearby amino acid could provide protection against reactive oxygen species [139].
Structural studies of the [FeFe]-hydrogenase maturation process are few, but Happe et al. have shown that incorporation of the [2Fe]H cluster is guided by arginine R449 and lysine L358 of CpI [140]. The molecular backbone movements could be studied by dipolar-dipolar EPR spectroscopy, e.g., between the H-cluster and a nitroxide label or between multiple nitroxide labels that have been incorporated by site-directed mutagenesis [141]. The structure of CrHydA1 apo-protein is controversial as the ‘open’ fold identified by Peters et al. significantly differs from holo-enzyme [67], while Happe et al. reported a structure that is barely any different from functional CrHydA1 [80]. Structural in situ data on the process of maturation will help solving the remaining questions.
To date, quantum chemical calculations have difficulties in predicting the principal values of the g-tensor and are only used to qualitatively assign spin densities when simulating interactions with surrounding nuclei [123, 125]. As DFT and atomic orbital quantum chemical computing techniques become more accurate for open shell systems, such as metalloenzymes, experimental data of full g- and hyperfine-tensors will be required to benchmark these simulations [142, 143]. However, the techniques described above rely on orientation selection of the frozen solution. To this end, one can either increase the operating frequency of the EPR experiment (separating the gxx, gyy, and gzz components) or perform single-crystal EPR experiments. Single-crystal EPR experiments are the ultimate method in determining the full magnetic interactions (magnitude and orientation), reflecting the electronic structure of the unpaired electron at an active site of an enzyme, and relating the magnetic principal axes to the proposed protein structure. Unlike [NiFe]-hydrogenase, which produced crystal dimensions on the order of 1 × 0.5 × 0.5 mm3 (250 nl), the application of single-crystal EPR to [FeFe]-hydrogenase was severely limited by the typically small crystal sizes with volumes in the nanoliter to sub-nanoliter range (0.3 × 0.1 × 0.1 mm3; 3 nl). In general, this limitation, due to the size and general flexibility of proteins, makes growing large crystals problematic [144]. Technical developments for EPR have allowed, for the first time, measurement of the [2Fe]H subcluster in the Hox state within a 3 nl protein crystal to be measured and the g-tensor proposed [92]. Further experiments to measure the hyperfine couplings are required to better evaluate DFT EPR calculations. Of course, the whole toolset of isotope doping can be performed if single crystals form.

Vibrational spectroscopy
The term ‘vibrational spectroscopy’ comprises two main techniques: Fourier-transform infrared (FTIR) spectroscopy and Raman spectroscopy. FTIR spectroscopy probes the frequency of molecules that undergo a distance-dependent change in dipole moment, whereas bonds with a vibrational change in polarizability are investigated by Raman spectroscopy most conveniently. The intense absorbance of liquid water and the amide bonds of the protein backbone (vC=O, vN–H) render FTIR spectroscopy a key technique in protein research [145]; however, probing metal centers directly, Raman spectroscopy is more powerful [146]. Lastly, nuclear resonance vibrational spectroscopy (NRVS) is an X-ray technique that exploits the recoil fraction of Mössbauer absorption, e.g., in 57Fe-labeled samples of hydrogenase [147,148,149], resulting in sidebands that report on vibrations that are coupled to the mental centers.
Characterization of different H-cluster intermediates
The active site cofactor of [FeFe]-hydrogenase carries CN– and CO ligands, which was first rationalized as a common feature among hydrogenase by Albracht et al. in 1996 [53]. The stretching frequency of these groups is observed in a regime devoid of (strong) water or protein bands, and the absorption coefficient of both CN– and CO is very high. Therefore, FTIR spectroscopy is a valuable tool investigating the H-cluster directly [150]. Following the publication of the XRD structures of DdHydAB and CpI, FTIR spectroscopy was exploited to characterize Hox, Hred, and Hox-CO [151,152,153]. The lack of a low-frequency µCO band in Hred (whose spectrum qualitatively resembles Hsred, compare Fig. 9) appeared to match the ‘semi-bridging’ CO ligand in the reduced XRD structure of DdHydAB [60].

Fits for the FTIR spectra of the same H-cluster states as in Fig. 7, additionally including the diamagnetic states Hred´, Hred, and Htrans. Characteristic frequencies are annotated as observed in DdHydAB (Htrans/Hinact, marked with an asterisk) and CrHydA1 (everything else)
Things became more complicated when CrHydA1 was analyzed by FTIR spectro-electrochemistry in 2009 [154]. The observation of a µCO band under reducing conditions was interpreted to hint at an isomer of Hred carrying a bridging CO ligand, seemingly explaining how the enzyme stabilizes the ‘rotated geometry’ of the diiron site [155,156,157]. Such ligand was absent in Hsred, first observed as a minor species in DdHydAB [107] and thoroughly characterized in CrHydA1 in 2012 [158]. Investigating CrHydA1 by resonance Raman spectroscopy and FTIR spectroscopy, various authors reported that the µCO band of Hred must be assigned to another one-electron reduced state, Hred´ [34, 159]. This intermediate, reduced at the [4Fe]H cluster, has been observed in H-cluster variant PDT earlier, where the central amine of the ADT ligand is replaced by a propane head group that efficiently prevents protonation and reduction of the diiron site [119]. Figure 10A highlights how small energetic differences (< 10 cm−1) between Hox/Hred´ and Hred/Hsred reflect the redox state of the [4Fe]H cluster while redox changes at the [2Fe]H site result in larger shifts (> 50 cm−1). The conversion from Hox into Hred includes an additional change in geometry: upon reduction, the Fe–Fe distance increases by 0.15 Å [160], the µCO ligand is liberated and ends up in a terminal position. Systematically comparing different models of the Hred or Hsred states, the IR pattern in Fig. 9 represents an H-cluster intermediate with a bridging hydride (µH–) most likely [161]. Stripp and Haumann have argued that such intermediates are not part of the catalytic cycle of hydrogen turnover but might play a role in ‘regulatory’ [FeFe]-hydrogenases [25], which often adopt Hred as a quasi-resting state [162, 163]. Interestingly, FTIR spectroscopy has shown that the CO ligand can be arrested in Fe–Fe bridging position upon reduction at cryogenic temperatures [94], giving rise to redox states HredH+ and HsredH+, presumably protonated at the ADT ligand [164, 165]. Due to the lack of direct experimental evidence for a µH– ligand in Hred/Hsred or +NH2 in HredH+/HsredH+, the controversy whether these states are catalytically relevant maintains [25,26,27]. We will return to a brief discussion of the catalytic cycle at the end of this section.

A FTIR difference spectra of the electrochemical reduction of CrHydA1 at pH 9 (‘Hred´–Hox’) and pH 6 (‘Hsred–Hred’). The small band shifts result from redox changes at the [4Fe]H cluster (only the strong Fed–CO bands are annotated). Note that the H-cluster lacks a µCO signal in the diiron site-reduced states Hred and Hsred. The band at 1972 cm−1 (*) remains unassigned. B FTIR difference spectra of the reaction of cofactor variant CrHydA1ODT with H2 (H2O) or D2 (D2O) in the CO regime of the H-cluster. Negative and positive bands are assigned to Hox and Hhyd, respectively. While none of the terminal CO ligands are affected, the downshift of the µCO band from 1862 cm−1 (blue) to 1856 cm−1 (orange) upon deuteration proves the presence of a terminal hydride in Hhyd
While the EPR spectrum of Hhyd (Fig. 7) is in good agreement with an Htrans-like electronic structure ([4Fe]H+–[Fep(II)–Fed(II)]), vibrational spectroscopy was employed to identify the terminal hydride ligand (tH–) directly. Figure 10B shows an FTIR difference spectrum in the H-cluster’s CO regime that results from the reaction of CrHydA1 cofactor variant ODT with H2 (in the presence of H2O) or D2 (in the presence of D2O). In this semi-artificial enzyme, the natural NH head group is exchanged against oxygen [69], which leads to an inhibition of proton transfer and accumulation of Hhyd when reduced with hydrogen gas [166]. Upon reaction with D2 instead of H2, a specific downshift of the µCO band at 1862 cm−1 is observed (Fig. 10B) that has been explained by vibrational energy transfer between tH– and µCO [115]. The ‘deuteride’ ligand (tD–) shows no such ‘trans coupling’, therefore, the µCO vibration shifts to slightly lower energies. Investigating 57Fe-labeled CrHydA1ODT by NRVS the low-frequency Fe-H– bending modes have been identified and demonstrated to shift by ~ 100 cm−1 in deuterated samples [149]. This is direct proof for the presence of a terminal hydride ligand in Hhyd, a finding that was independently confirmed by NMR spectroscopy on CrHydA1 cysteine variant C169A [167].
These studies demonstrated an enrichment of Hhyd in different variants and established its FTIR fingerprint as a marker for amino acid residues relevant in catalytic proton transfer [55]. However, Stripp et al. were able to demonstrate the accumulation of Hhyd in wild-type [FeFe]-hydrogenase as well, reacting the enzymes with H2 at low pH [166]. Under these conditions, the oxidized starting state differs from Hox by a small shift to higher frequencies, which inspired us to propose an oxidized state with a protonated cysteine at the [4Fe]H cluster, HoxH [34]. The pH dependence of Em in the Hox/Hred´ conversion and the pH-dependent equilibrium between Hred and Hred´ under reducing conditions facilitated a model that requires proton transfer in order to stabilize the reduced [4Fe]H+ cluster [168,169,170]. Given the direct interconversion of HoxH and Hhyd, the same logic applies to the hydride state [166].
The catalytic cycle
Similar to the question whether the reduced diiron site is protonated at the ADT ligand (HredH+ and HsredH+) or in Fe–Fe bridging position (Hred and Hsred), direct evidence for proton transfer (PT) toward the [4Fe]H cluster in the formation of Hred´ is missing. In fact, Birrell et al. were not able to reproduce the pH dependence of the Hox/Hred´ transition [32], suggesting a simple electron transfer (ET) step rather than proton-coupled electron transfer (PCET). The different interpretations of the catalytic mechanism of [FeFe]-hydrogenase can be illustrated in a simplified cycle comprising of either five steps or three steps (Fig. 11).

Catalytic mechanism of [FeFe]-hydrogenase including the five main H-cluster states. The cubane represents the [4Fe]H cluster in the oxidized (+ 2) or reduced state (+ 1). The rectangular box depicts the diiron site in the mixed-valence (I/II), reduced (I/I), and ‘super oxidized’ state (II/II). {H} and (H) refer to protonation of the [4Fe]H cluster and diiron site, respectively. See text for details on the ‘five-step cycle’ (ABCDE) and the ‘three-step cycle’ (A*B*E)
In H2 evolution direction, the ‘five-step model’ starts with external reduction of [4Fe]H cluster (A), followed by intramolecular ET from [4Fe]H to the diiron site and protonation of the ADT ligand (B). This step is rationalized by the assumption that a reduction of the diiron site does not change the µCO geometry of the H-cluster [164], ruling out the formation of a bridging hydride. A chemically reasonable explanation for the observed pH dependence [165] would be protonation of the ADT ligand, hence we are using the HredH+ nomenclature here. External re-reduction of [4Fe]H produces HsredH+ (C) before PCET from the ADT ligand to the distal iron ion triggers the formation of Hhyd (D). The later step, hypothetical for a long time, has been demonstrated by cryogenic FTIR in photochemical experiments in 2020 [117]. With a second proton coming in, the hydride ligand combines to H2, which restores the Hox state (E). The three-step model starts with PCET in the formation of Hred´ (A*). Note that the proton {H} is not part of the hydrogen turnover reaction but binds at the [4Fe]H cluster to impede intramolecular ET to the diiron site. In a second step of PCET, Hred´ is converted into Hhyd directly (B*) before H2 is released upon protonation of the hydride ligand as described above (E), either recovering Hox (when {H} is released from the [4Fe]H cluster) or HoxH. The later state is observed under turnover conditions at low pH exclusively [170].
Future directions
The strong and distinct IR absorption of the H-cluster’s CO and CN– ligands has facilitated time-resolved ‘flash photolysis’ experiments using quantum cascade laser (QCL) radiation [171,172,173]. Within hundreds of µs, Dyer et al. investigated the kinetic competence of various reduced H-cluster states concluding that, “the rate limiting step of catalysis is not the interconversion of any of the known intermediate states” [172]. Understanding hydrogen turnover with [FeFe]-hydrogenase hence means understanding the electronic properties of the H-cluster rather than the redox kinetics. To this end, ultrafast two-dimensional infrared (2DIR) spectroscopy may help [174]. Using this technique, the vibrational excitation of single ligands and energy transfer among ligands can be probed without the need for isotope editing [175], as demonstrated by Horch et al. at the example of [NiFe]-hydrogenase [176,177,178]. Similar experiments have not been published on [FeFe]-hydrogenase yet; however, an application can be expected in the future.
Moreover, 2DIR spectroscopy is a technique used to probe the coupling between cofactor and protein environment, identifying structural or electrostatic changes in the second coordination sphere. Such data are included in difference spectra ‘beyond’ the energy regime of the active site H-cluster. In [NiFe]- and [FeFe]-hydrogenase, in situ FTIR difference spectroscopy helped understanding proton transfer between active site and solvent [35], highlighting the role of SH- and COOH-containing amino acids, and water molecules [56, 179]. Generating difference spectra is severely complicated when light is excluded as a trigger. While certainly a helpful tool, visible light irradiation affects the H-cluster in various ways [117, 175] and must be handled with care. Gas titrations present a unique way to trigger the reactivity of hydrogenases and generate ATR FTIR difference spectra [150]. Such experiment may elucidate the process of oxidative H-cluster inactivation, CO inhibition, or H2 binding.
While biophysical experiments are mostly performed in condensed phase, structural data with atomistic resolution is obtained from XRD on protein crystals. Bridging this gap, FTIR microspectroscopy and FTIR spectro-electrochemistry has been used to probe [NiFe]-hydrogenase crystals [180, 181] and [FeFe]-hydrogenase crystals [93]. In the future, we expect that FTIR spectroscopy and EPR spectroscopy (92) will be used on hydrogenase crystals routinely.

Data availability
The datasets analysed during the current study are available from the corresponding authors on request.
References
Greening C, Grinter R (2022) Microbial oxidation of atmospheric trace gases. Nat Rev Microbiol 20:513–528
Schwartz E, Friedrich B (2006) The H2-metabolizing prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, ed. 3. Springer, New York, pp. 496–563. https://link.springer.com/referenceworkentry/10.1007%2F0-387-30742-7_17),
Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, Cook GM, Morales SE (2016) Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J 10:761–777
Benoit SL, Maier RJ, Sawers RG, Greening C (2020) Molecular hydrogen metabolism: a widespread trait of pathogenic bacteria and protists. Microbiol Mol Biol Rev 84:e00092-e119
Das D, Veziroǧlu TN (2001) Hydrogen production by biological processes: a survey of literature. Int J Hydrogen Energy 26:13–28
Tamagnini P, Axelsson R, Lindberg P, Oxelfelt F, Wünschiers R, Lindblad P (2002) Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol Mol Biol Rev 66:1–20
Melis A, Happe T (2004) Trails of green alga hydrogen research—from hans gaffron to new frontiers. Photosynth Res 80:401–409
Vignais PM, Billoud B (2007) Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272
Lubitz W, Ogata H, Rüdiger O, Reijerse E, Rudiger O, Reijerse E (2014) Hydrogenases. Chem Rev 114:4081–4148
Søndergaard D, Pedersen CNS, Greening C (2016) HydDB: a web tool for hydrogenase classification and analysis. Sci Rep 6:34212
Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U (2008) The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science (80–). 321:572–575
Thauer RK, Kaster A-K, Goenrich M, Schick M, Hiromoto T, Shima S (2010) Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem 79:507–536
Huang G, Wagner T, Ermler U, Shima S (2020) Methanogenesis involves direct hydride transfer from H2 to an organic substrate. Nat Rev Chem 4:213–221
Bothe H, Schmitz O, Yates MG, Newton WE (2010) Nitrogen fixation and hydrogen metabolism in cyanobacteria. Microbiol Mol Biol Rev 74:529–551
Fritsch J, Lenz O, Friedrich B (2013) Structure, function and biosynthesis of O2-tolerant hydrogenases. Nat Rev 11:106–114
Parkin A, Sargent F (2012) The hows and whys of aerobic H2 metabolism. Curr Opin Chem Biol 16:26–34
Shafaat HS, Rüdiger O, Ogata H, Lubitz W (2013) [NiFe] hydrogenases: a common active site for hydrogen metabolism under diverse conditions. Biochim Biophys Acta 1827:986–1002
Armstrong FA, Fontecilla-Camps JC (2008) A natural choice for activating hydrogen. Science (80–). 321:498–499
Schuchmann K, Chowdhury NP, Müller V (2018) Complex multimeric [FeFe] hydrogenases: biochemistry, physiology and new opportunities for the hydrogen economy. Front Microbiol 9:2911
Goldet G, Brandmayr C, Stripp ST, Happe T, Cavazza C, Fontecilla-Camps JC, Armstrong FA (2009) Electrochemical kinetic investigations of the reactions of [FeFe]-hydrogenases with carbon monoxide and oxygen: comparing the importance of gas tunnels and active-site electronic/redox effects. J Am Chem Soc 131:14979–14989
del Barrio M, Sensi M, Fradale L, Bruschi M, Greco C, de Gioia L, Bertini L, Fourmond V, Léger C (2018) Interaction of the H-cluster of FeFe hydrogenase with halides. J Am Chem Soc 140:5485–5492
Vincent KA, Parkin A, Armstrong FA (2007) Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem Rev 107:4366–4413
Armstrong FA, Evans RM, Hexter SV, Murphy BJ, Roessler MM, Wul P (2016) Guiding principles of hydrogenase catalysis instigated and clarified by protein film electrochemistry. Acc Chem Res 49:884–892
Fourmond V, Plumeré N, Léger C (2021) Reversible catalysis. Nat Rev Chem 5:348–360
Haumann M, Stripp ST (2018) The molecular proceedings of biological hydrogen turnover. Acc Chem Res 51:1755–1763
Kleinhaus JT, Wittkamp F, Yadav S, Siegmund D, Apfel U-P (2021) [FeFe]-Hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem Soc Rev 50:1668–1784
Birrell JA, Rodríguez-Maciá P, Reijerse EJ, Martini MA, Lubitz W (2021) The catalytic cycle of [FeFe] hydrogenase: a tale of two sites. Coord Chem Rev 449:214191
Land H, Senger M, Berggren G, Stripp ST (2020) Current state of [FeFe]-hydrogenase research—biodiversity and spectroscopic investigations. ACS Catal 10:7069–7086
Britt RD, Tao L, Rao G, Chen N, Wang L-P (2022) Proposed mechanism for the biosynthesis of the [FeFe] hydrogenase H-cluster: central roles for the radical SAM enzymes HydG and HydE. ACS Bio Med Chem Au 2:11–21
Stripp ST, Duffus BR, Fourmond V, Léger C, Leimkühler S, Hirota S, Hu Y, Jasniewski A, Ogata H, Ribbe MW (2022) Second and outer coordination sphere effects in nitrogenase, hydrogenase, formate dehydrogenase, and CO dehydrogenase. Chem Rev 122:11900–11973
Winkler M, Hemschemeier A, Jacobs J, Stripp ST, Happe T (2010) Multiple ferredoxin isoforms in Chlamydomonas reinhardtii - their role under stress conditions and biotechnological implications. Eur J Cell Biol 89:998–1004
Rodríguez-Maciá P, Breuer N, DeBeer S, Birrell JA (2020) Insight into the redox behavior of the [4Fe–4S] subcluster in [FeFe] hydrogenases. ACS Catal 10:13084–13095
Artz JH, Zadvornyy OA, Mulder DW, Keable SM, Cohen AE, Ratzloff MW, Williams SG, Ginovska B, Kumar N, Song J, McPhillips SE, Davidson CM, Lyubimov AY, Pence N, Schut GJ, Jones AK, Soltis SM, Adams MWW, Raugei S, King PW, Peters JW (2020) Tuning catalytic bias of hydrogen gas producing hydrogenases. J Am Chem Soc 142:1227–1235
Senger M, Mebs S, Duan J, Shulenina O, Laun K, Kertess L, Wittkamp F, Apfel U-P, Happe T, Winkler M, Haumann M, Stripp ST (2018) Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases. Phys Chem Chem Phys 20:3128–3140
Tai H, Hirota S, Stripp ST (2021) Proton transfer mechanisms in bimetallic hydrogenases. Acc Chem Res 54:232–241
Duan J, Mebs S, Laun K, Wittkamp F, Heberle J, Happe T, Hofmann E, Apfel U-P, Winkler M, Senger M, Haumann M, Stripp ST (2019) Geometry of the catalytic active site in [FeFe]-hydrogenase is determined by hydrogen bonding and proton transfer. ACS Catal 9:9140–9149
Lampret O, Adamska-Venkatesh A, Konegger H, Wittkamp F, Apfel U-P, Reijerse EJ, Lubitz W, Rüdiger O, Happe T, Winkler M (2017) Interplay between CN – ligands and the secondary coordination sphere of the H-cluster in [FeFe]-hydrogenases. J Am Chem Soc 139:18222–18230
Adams MW (1990) The structure and mechanism of iron-hydrogenases. Biochim Biophys Acta 1020:115–145
Stephenson M, Stickland LH (1931) Hydrogenase: a bacterial enzyme activating molecular hydrogen: the properties of the enzyme. Biochem J 25:205–214
Farkas A, Farkas L, Yudkin J (1934) The decomposition of sodium formate by bacterium coli in the presence of heavy water. Proc R Soc Lond Ser B Contain. Pap A Biol Character 115:373–379
Gest H (1954) Oxidation and evolution of molecular hydrogen by microorganisms. Bacteriol Rev 18:43–73
Peck HD, Pietro AS, Gest H (1956) On the mechanism of hydrogenase in action. Proc Natl Acad Sci 42:13–19
Sadana JC, Rittenberg D (1963) Some observations on the enzyme hydrogenase of Desulfovibrio desulfuricans. Proc Natl Acad Sci 50:900–904
Nakos G, Mortenson L (1971) Purification and properties of hydrogenase, an iron sulfur protein, from Clostridium pasteurianum W5. Biochim Biophys Acta 227:576–583
Mortenson LE, Nakos G (1971) Structural properties of hydrogenase from Clostridium pasteurianum W5. Biochemistry 10:2442–2449
Adams MWW, Mortenson LE, Chen J-S (1980) Hydrogenase. Biochim Biophys Acta Rev Bioenerg 594:105–176
Cammack R, Maharajh WVL, Schneider K (1982) EPR studies of some oxygen-stable hydrogenases. In: Ho C, Eaton WA, Collman JP, Gibson QH, Leigh JS, Margoliash E, Moffat K, Scheidt WR (eds) Electron transport and oxygen utilization,. Macmillan Education London, pp. 411–415. https://doi.org/10.1007/978-1-349-06491-5)
Yamazaki S (1982) A selenium-containing hydrogenase from Methanococcus vannielii. Identification of the selenium moiety as a selenocysteine residue. J Biol Chem 257:7926–7929
Graf E-G, Thauer RK (1981) Hydrogenase from methanobacterium thermoautotrophicum, a nickel-containing enzyme. FEBS Lett 136:165–169
Zirngibl C, Dongen W, Schworer B, Bunau R, Richter M, Klein A, Thauer RK (1992) H2-forming methylenetetrahydromethanopterin dehydrogenase, a novel type of hydrogenase without iron-sulfur clusters in methanogenic archaea. Eur J Biochem 208:511–520
Volbeda A, Charon M, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC (1995) Crystal structure of the nickel–iron hydrogenase from Desulfovibrio desulfuricans. Nature 373:580–587
Happe RP, Roseboom W, Pierik AJ, Albracht SPJ, Bagley KA (1997) Biological activition of hydrogen. Nature 385:126–126
van der Spek TM, Arendsen AF, Happe RP, Yun S, Bagley KA, Stufkens DJ, Hagen WR, Albracht SPJ, Spek TM, Arendsen AF, Happe RP, Yun S, Bagley KA, Stufkens DJ, Hagen WR, Albracht SPJ, Hagen WR (1996) Similarities in the architecture of the active sites of Ni-hydrogenases and Fe-hydrogenases detected by means of infrared spectroscopy. Eur J Biochem 237:629–634
Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC (1998) X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution. Science 282:1853–1858
Duan J, Senger M, Esselborn J, Engelbrecht V, Wittkamp F, Apfel U-P, Hofmann E, Stripp ST, Happe T, Winkler M (2018) Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases. Nat Commun 9:4726
Senger M, Eichmann V, Laun K, Duan J, Wittkamp F, Knör G, Apfel U-P, Happe T, Winkler M, Heberle J, Stripp ST (2019) How [FeFe]-hydrogenase facilitates bidirectional proton transfer. J Am Chem Soc 141:17394–17403
Lampret O, Duan J, Hofmann E, Winkler M, Armstrong FA, Happe T (2020) The roles of long-range proton-coupled electron transfer in the directionality and efficiency of [FeFe]-hydrogenases. Proc Natl Acad Sci 117:20520–20529
Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC (1999) Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 7:13–23
Lemon BJ, Peters JW (2000) Photochemistry at the active site of the carbon monoxide inhibited form of the iron-only hydrogenase (CpI). J Am Chem Soc 122:3793–3794
Nicolet Y, de Lacey AL, Vernède X, Fernandez VM, Hatchikian EC, Fontecilla-Camps JC (2001) Crystallographic and FTIR spectroscopic evidence of changes in fe coordination upon reduction of the active site of the Fe-Only hydrogenase from Desulfovibrio desulfuricans. J Am Chem Soc 123:1596–1601
Stripp ST, Happe T (2009) How algae produce hydrogen - news from the photosynthetic hydrogenase. Dalt Trans 45:9960–9969
Happe T, Naber JD (1993) Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii. Eur J Biochem 214:475–481
von Abendroth G, Stripp ST, Silakov A, Croux C, Soucaille P, Girbal L, Happe T (2008) Optimized over-expression of [FeFe] hydrogenases with high specific activity in Clostridium acetobutylicum. Int J Hydrogen Energy 33:6076–6081
Sybirna K, Antoine T, Lindberg P, Fourmond V, Rousset M, Méjean V, Bottin H (2008) Shewanella oneidensis: a new and efficient System for expression and maturation of heterologous [Fe-Fe] Hydrogenase from Chlamydomonas reinhardtii. BMC Biotechnol 8:73
Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M, Ghirardi ML (2004) Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active [Fe] hydrogenase. J Biol Chem 279:25711–25720
King PW, Posewitz MC, Ghirardi ML, Seibert M (2006) Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. J Bacteriol 188:2163
Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW (2010) Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA(DeltaEFG). Nature 465:248–251
Happe T, Kaminski A (2002) Differential regulation of the Fe-hydrogenase during anaerobic adaptation in the green alga Chlamydomonas reinhardtii. Eur J Biochem 269:1022–1032
Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca J-M, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M (2013) Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 499:66–69
Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T (2013) Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat Chem Biol 9:607–609
Birrell JA, Wrede K, Pawlak K, Rodriguez-Maciá P, Rüdiger O, Reijerse EJ, Lubitz W (2016) Artificial maturation of the highly active heterodimeric [FeFe] hydrogenase from Desulfovibrio desulfuricans ATCC 7757. Isr J Chem 56:852–863
Adamska-Venkatesh A, Simmons TR, Siebel JF, Artero V, Fontecave M, Reijerse E, Lubitz W (2015) Artificially maturated [FeFe] hydrogenase from Chlamydomonas reinhardtii: a HYSCORE and ENDOR study of a non-natural H-cluster. Phys Chem Chem Phys 17:5421–5430
Adamska-Venkatesh A, Roy S, Siebel JF, Simmons TR, Fontecave M, Artero V, Reijerse E, Lubitz W (2015) Spectroscopic characterization of the bridging amine in the active site of [FeFe] hydrogenase using isotopologues of the H-Cluster. J Am Chem Soc 137:12744–12747
Gilbert-Wilson R, Siebel JF, Adamska-Venkatesh A, Pham CC, Reijerse E, Wang H, Cramer SP, Lubitz W, Rauchfuss TB (2015) Spectroscopic investigations of [FeFe] hydrogenase maturated with [57Fe2(adt)(CN)2(CO)4]2-. J Am Chem Soc 137:8998–9005
Siebel JF, Adamska-Venkatesh A, Weber K, Rumpel S, Reijerse E, Lubitz W (2015) Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry 54:1474–1483
Sommer C, Richers CP, Lubitz W, Rauchfuss TB, Reijerse EJ (2018) A [RuRu] analogue of an [FeFe]-hydrogenase traps the key hydride intermediate of the catalytic cycle. Angew Chemie Int Ed 57:5429–5432
Sommer C, Rumpel S, Roy S, Farès C, Artero V, Fontecave M, Reijerse E, Lubitz W (2018) Spectroscopic investigations of a semi-synthetic [FeFe] hydrogenase with propane di-selenol as bridging ligand in the binuclear subsite: comparison to the wild type and propane di-thiol variants. JBIC J Biol Inorg Chem 23:481–491
Noth J, Esselborn J, Güldenhaupt J, Brünje A, Sawyer A, Apfel U-P, Gerwert K, Hofmann E, Winkler M, Happe T (2016) [FeFe]-hydrogenase with chalcogenide substitutions at the H-cluster maintains Full H2 evolution activity. Angew Chemie Int Ed 55:8396–8400
Kertess L, Wittkamp F, Sommer C, Esselborn J, Rüdiger O, Reijerse EJ, Hofmann E, Lubitz W, Winkler M, Happe T, Apfel U-P (2017) Chalcogenide substitution in the [2Fe] cluster of [FeFe]-hydrogenases conserves high enzymatic activity. Dalt Trans 46:16947–16958
Esselborn J, Muraki N, Klein K, Engelbrecht V, Metzler-Nolte N, Apfel U-P, Hofmann E, Kurisu G, Happe T (2016) A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem Sci 7:959–968
Fritsch J, Scheerer P, Frielingsdorf S, Kroschinsky S, Friedrich B, Lenz O, Spahn CMT (2011) The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 479:249–252
Frielingsdorf S, Fritsch J, Schmidt A, Hammer M, Löwenstein J, Siebert E, Pelmenschikov V, Jaenicke T, Kalms J, Rippers Y, Lendzian F, Zebger I, Teutloff C, Kaupp M, Bittl R, Hildebrandt P, Friedrich B, Lenz O, Scheerer P (2014) Reversible [4Fe-3S] cluster morphing in an O2-tolerant [NiFe] hydrogenase. Nat Chem Biol 10:378–385
Stripp ST, Goldet G, Brandmayr C, Sanganas O, Vincent KA, Haumann M, Armstrong FA, Happe T (2009) How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc Natl Acad Sci USA 106:17331–17336
Swanson KD, Ratzloff MW, Mulder DW, Artz JH, Ghose S, Hoffman A, White S, Zadvornyy OA, Broderick JB, Bothner B, King PW, Peters JW (2015) [FeFe]-hydrogenase oxygen inactivation is initiated at the H cluster 2Fe subcluster. J Am Chem Soc 137:1809–1816
Esselborn J, Kertess L, Apfel U-P, Hofmann E, Happe T (2019) Loss of Specific active-site iron atoms in oxygen-exposed [FeFe]-hydrogenase determined by detailed X-ray structure analyses. J Am Chem Soc 141:17721–17728
Mebs S, Kositzki R, Duan J, Kertess L, Senger M, Wittkamp F, Apfel U-P, Happe T, Stripp ST, Winkler M, Haumann M (2017) Hydrogen and oxygen trapping at the H-cluster of [FeFe]-hydrogenase revealed by site-selective spectroscopy and QM/MM calculations. BBA Bioenerg 1859:28–41
Winkler M, Duan J, Rutz A, Felbek C, Scholtysek L, Lampret O, Jaenecke J, Apfel U-P, Gilardi G, Valetti F, Fourmond V, Hofmann E, Léger C, Happe T (2021) A safety cap protects hydrogenase from oxygen attack. Nat Commun 12:756
Corrigan PS, Tirsch JL, Silakov A (2020) Investigation of the unusual ability of the [FeFe] hydrogenase from clostridium beijerinckii to access an O2-protected state. J Am Chem Soc 142:12409–12419
Rodríguez-Maciá P, Reijerse EJ, Van Gastel M, Debeer S, Lubitz W, Rüdiger O, Birrell JA (2018) Sulfide protects [FeFe] hydrogenases from O2. J Am Chem Soc 140:9346–9350
Felbek C, Arrigoni F, de Sancho D, Jacq-Bailly A, Best RB, Fourmond V, Bertini L, Léger C (2021) Mechanism of hydrogen sulfide-dependent inhibition of FeFe hydrogenase. ACS Catal 11:15162–15176
Rodríguez-Maciá P, Galle LM, Bjornsson R, Lorent C, Zebger I, Yoda Y, Cramer SP, DeBeer S, Span I, Birrell JA (2020) Caught in the hinact: crystal structure and spectroscopy reveal a sulfur bound to the active site of an O2-stable state of [FeFe] hydrogenase. Angew Chemie Int Ed 59:16786–16794
Sidabras JW, Duan J, Winkler M, Happe T, Hussein R, Zouni A, Suter D, Schnegg A, Lubitz W, Reijerse EJ (2019) Extending electron paramagnetic resonance to nanoliter volume protein single crystals using a self-resonant microhelix. Sci Adv eaay5:1394
Morra S, Duan J, Winkler M, Ash PA, Happe T, Vincent KA (2021) Electrochemical control of [FeFe]-hydrogenase single crystals reveals complex redox populations at the catalytic site. Dalt Trans 50:12655–12663
Stripp ST, Mebs S, Haumann M (2020) Temperature dependence of structural dynamics at the catalytic cofactor of [FeFe]-hydrogenase. Inorg Chem 59:16474–16488
Furlan C, Chongdar N, Gupta P, Lubitz W, Ogata H, Blaza JN, Birrell JA (2022) Structural insight on the mechanism of an electron-bifurcating [FeFe] hydrogenase. Elife 11:e79361
Katsyv A, Kumar A, Saura P, Pöverlein MC, Freibert SA, Stripp ST, Jain S, Gamiz-Hernandez AP, Kaila VRI, Müller V, Schuller JM (2023) Molecular basis of the electron bifurcation mechanism in the [FeFe]-hydrogenase complex HydABC. J Am Chem Soc. https://doi.org/10.1021/jacs.2c11683
Hiromoto T, Nishikawa K, Inoue S, Matsuura H, Hirano Y, Kurihara K, Kusaka K, Cuneo M, Coates L, Tamada T, Higuchi Y (2020) Towards cryogenic neutron crystallography on the reduced form of [NiFe]-hydrogenase. Acta Crystallogr Sect D Struct Biol 76:946–953
Eisermann J, Seif-Eddine M, Roessler MM (2021) Insights into metalloproteins and metallodrugs from electron paramagnetic resonance spectroscopy. Curr Opin Chem Biol 61:114–122
Hagen WR (2018) EPR spectroscopy of complex biological iron–sulfur systems. J Biol Inorg Chem 23:623–634
Lubitz W, Reijerse E, van Gastel M (2007) [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem Rev 107:4331–4365
Patil DS, Moura JJ, He SH, Teixeira M, Prickril BC, DerVartanian DV, Peck HD, LeGall J, Huynh BH (1988) EPR-detectable redox centers of the periplasmic hydrogenase from Desulfovibrio vulgaris. J Biol Chem 263:18732–18738
Pierik AJ, Hagen WR, Redeker JS, Wolbert RBG, Boersma M, Verhagen MFJM, Grande HJ, Veeger C, Mutsaers PHA, Sands RH, Dunham WR (1992) Redox properties of the iron-sulfur clusters in activated Fe-hydrogenase from Desulfovibrio vulgaris (Hildenborough). Eur J Biochem 209:63–72
Bennett B, Lemon BJ, Peters JW (2000) Reversible carbon monoxide binding and inhibition at the active site of the Fe-only hydrogenase. Biochemistry 39:7455–7460
Artz JH, Mulder DW, Ratzloff MW, Lubner CE, Zadvornyy OA, LeVan AX, Williams SG, Adams MWW, Jones AK, King PW, Peters JW (2017) Reduction potentials of [FeFe]-hydrogenase accessory iron-sulfur clusters provide insights into the energetics of proton reduction catalysis. J Am Chem Soc 139:9544–9550
Rodríguez-Maciá P, Pawlak K, Rüdiger O, Reijerse EJ, Lubitz W, Birrell JA (2017) Intercluster redox coupling influences protonation at the H-cluster in [FeFe] hydrogenases. J Am Chem Soc 139:15122–15134
Albracht SPJ, Roseboom W, Hatchikian EC (2006) The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. I. Light sensitivity and magnetic hyperfine interactions as observed by electron paramagnetic resonance. J Biol Inorg Chem 11:88–101
Roseboom W, De Lacey AL, Fernandez VM, Hatchikian EC, Albracht SPJ (2006) The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy. J Biol Inorg Chem 11:102–118
Silakov A, Reijerse EJ, Lubitz W (2011) Unraveling the electronic properties of the photoinduced states of the H-cluster in the [FeFe] hydrogenase from D. desulfuricans. Eur J Inorg Chem 2011:1056–1066
Rusnak FM, Adams MWW, Mortenson LE, Münck E (1987) Mössbauer study of Clostridium pasteurianum hydrogenase II. Evidence for a novel three-iron cluster. J Biol Chem 262:38–41
Popescu C, Münck E (1999) Electronic structure of the H cluster in [FeFe]-hydrogenases. J Am Chem Soc 121:15054–15061
Pereira ASS, Tavares P, Moura I, Moura JJGJ, Huynh BHH (2001) Mössbauer characterization of the iron-sulfur clusters in desulfovibrio v ulgaris hydrogenase. J Am Chem Soc 123:2771–2782
Duan J, Hemschemeier A, Burr DJ, Stripp ST, Hofmann E, Happe T (2023) Cyanide binding to [FeFe]-hydrogenase stabilizes the alternative configuration of the proton transfer pathway. Angew Chemie Int Ed. https://doi.org/10.1002/anie.202216903
Martini MA, Bikbaev K, Pang Y, Lorent C, Wiemann C, Breuer N, Zebger I, DeBeer S, Span I, Bjornsson R, Birrell JA, Rodríguez-Maciá P (2023) Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases. Chem Sci. https://doi.org/10.1039/D2SC06098A
Mulder DW, Ratzloff MW, Shepard EM, Byer AS, Noone SM, Peters JW, Broderick JB, King PW (2013) EPR and FTIR analysis of the mechanism of H2 activation by [FeFe]-hydrogenase HydA1 from Chlamydomonas reinhardtii. J Am Chem Soc 135:6921–6929
Mulder DW, Ratzloff MW, Bruschi M, Greco C, Koonce E, Peters JW, King PW (2014) Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase. J Am Chem Soc 136:15394–15402
Mulder DW, Guo Y, Ratzloff MW, King PW (2017) Identification of a catalytic iron-hydride at the H-cluster of [FeFe]-hydrogenase. J Am Chem Soc 139:83–86
Lorent C, Katz S, Duan J, Kulka CJ, Caserta G, Teutloff C, Yadav S, Apfel U-P, Winkler M, Happe T, Horch M, Zebger I (2020) Shedding light on proton and electron dynamics in [FeFe] hydrogenases. J Am Chem Soc 142:5493–5497
Sevdalina L, Thorsten M, Klaus Z, Ulrich B, Bernd L, Thomas P (2010) Multifrequency pulsed electron paramagnetic resonance on metalloproteins. Acc Chem Res 43:181–189
Adamska-Venkatesh A, Krawietz D, Siebel JF, Weber K, Happe T, Reijerse E, Lubitz W (2014) New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster. J Am Chem Soc 136:11339–11346
Silakov A, Reijerse EJ, Albracht SPJ, Hatchikian EC, Lubitz W (2007) The electronic structure of the H-cluster in the [FeFe]-hydrogenase from Desulfovibrio desulfuricans: a Q-band 57Fe-ENDOR and HYSCORE study. J Am Chem Soc 129:11447–11458
Rao G, Britt RD (2018) Electronic structure of two catalytic states of the [FeFe] hydrogenase H-cluster as probed by pulse electron paramagnetic resonance spectroscopy. Inorg Chem 57:10935–10944
Myers WK, Stich TA, Suess DLM, Kuchenreuther JM, Swartz JR, Britt RD (2014) The cyanide ligands of [FeFe] hydrogenase: pulse EPR studies of 13C and 15N-labeled H-cluster. J Am Chem Soc 136:12237–12240
Fiedler AT, Brunold TC (2005) Computational studies of the H-cluster of Fe-only hydrogenases: geometric, electronic, and magnetic properties and their dependence on the [Fe4S4] cubane. Inorg Chem 44:9322–9334
Silakov A, Wenk B, Reijerse E, Albracht SPJ, Lubitz W (2009) Spin distribution of the H-cluster in the Hox-CO state of the [FeFe] hydrogenase from Desulfovibrio desulfuricans: HYSCORE and ENDOR study of 14N and 13C nuclear interactions. JBIC J Biol Inorg Chem 14:301–313
Greco C, Silakov A, Bruschi M, Ryde U, De Gioia L, Lubitz W (2011) Magnetic properties of [FeFe]-hydrogenases: a theoretical investigation based on extended QM and QM/MM models of the h-cluster and its surroundings. Eur J Inorg Chem 2011:1043–1049
Reijerse EJ, Pelmenschikov V, Birrell JA, Richers CP, Kaupp M, Rauchfuss TB, Cramer SP, Lubitz W (2019) Asymmetry in the ligand coordination sphere of the [FeFe] hydrogenase active site is reflected in the magnetic spin interactions of the Aza-propanedithiolate Ligand. J Phys Chem Lett 10:6794–6799
Reijerse E, Birrell JA, Lubitz W (2020) Spin polarization reveals the coordination geometry of the [FeFe] hydrogenase active site in Its CO-inhibited state. J Phys Chem Lett 11:4597–4602
Németh B, Senger M, Redman HJ, Ceccaldi P, Broderick J, Magnuson A, Stripp ST, Haumann M, Berggren G (2020) [FeFe]-hydrogenase maturation: H-cluster assembly intermediates tracked by electron paramagnetic resonance, infrared, and X-ray absorption spectroscopy. J Biol Inorg Chem 25:777–788
Laun K, Mebs S, Duan J, Wittkamp F, Apfel U-P, Happe T, Winkler M, Haumann M, Stripp ST (2018) Spectroscopical investigations on the redox chemistry of [FeFe]-hydrogenases in the presence of carbon monoxide. Molecules 23:1669
Rao G, Tao L, Suess DLM, Britt RD (2018) A [4Fe-4S]-Fe(CO)(CN)-l-cysteine intermediate is the first organometallic precursor in [FeFe] hydrogenase H-cluster bioassembly. Nat Chem 10:555–560
Britt RD, Rao G, Tao L (2020) Biosynthesis of the catalytic H-cluster of [FeFe] hydrogenase: the roles of the Fe–S maturase proteins HydE, HydF, and HydG. Chem Sci 11:10313–10323
Suess DLM, Britt RD (2015) EPR spectroscopic studies of [FeFe]-hydrogenase maturation. Top Catal 58:699–707
Rao G, Pattenaude SA, Alwan K, Blackburn NJ, Britt RD, Rauchfuss TB (2019) The binuclear cluster of [FeFe] hydrogenase is formed with sulfur donated by cysteine of an [Fe(Cys)(CO)2(CN)] organometallic precursor. Proc Natl Acad Sci USA 116:20850–20855
Rao G, Chen N, Marchiori DA, Wang L-P, Britt RD (2022) Accumulation and pulse electron paramagnetic resonance spectroscopic investigation of the 4-oxidobenzyl radical generated in the radical S -adenosyl- l -methionine Enzyme HydG. Biochemistry 61:107–116
Tao L, Pattenaude SA, Joshi S, Begley TP, Rauchfuss TB, Britt RD (2020) Radical SAM enzyme HydE generates adenosylated Fe(I) intermediates en route to the [FeFe]-hydrogenase catalytic H-cluster. J Am Chem Soc 142:10841–10848
Zhang Y, Tao L, Woods TJ, Britt RD, Rauchfuss TB (2022) Organometallic Fe2(μ-SH)2(CO)4(CN)2 cluster allows the biosynthesis of the [FeFe]-hydrogenase with only the hydf maturase. J Am Chem Soc 144:1534–1538
Pagnier A, Balci B, Shepard EM, Broderick WE, Broderick JB (2022) [FeFe]-Hydrogenase in vitro maturation. Angew Chemie Int Ed. https://doi.org/10.1002/anie.202212074
Heghmanns M, Rutz A, Kutin Y, Engelbrecht V, Winkler M, Happe T, Kasanmascheff M (2022) The oxygen-resistant [FeFe]-hydrogenase CbA5H harbors an unknown radical signal. Chem Sci. https://doi.org/10.1039/D2SC00385F
Colovic MB, Vasic VM, Djuric DM, Krstic DZ (2018) Sulphur-containing amino acids: protective role against free radicals and heavy metals. Curr Med Chem 25:324–335
Lampret O, Esselborn J, Haas R, Rutz A, Booth RL, Kertess L, Wittkamp F, Megarity CF, Armstrong FA, Winkler M, Happe T (2019) The final steps of [FeFe]-hydrogenase maturation. Proc Natl Acad Sci 116:15802–15810
Motion CL, Lovett JE, Bell S, Cassidy SL, Cruickshank PAS, Bolton DR, Hunter RI, El Mkami H, Van Doorslaer S, Smith GM (2016) DEER sensitivity between iron centers and nitroxides in heme-containing proteins improves dramatically using broadband. High-Field EPR J Phys Chem Lett 7:1411–1415
Neese F (2017) Quantum chemistry and EPR parameters, vol. 6. In: eMagRes. Wiley, Chichester, pp. 1–22. https://doi.org/10.1002/9780470034590.emrstm1505)
Tran VA, Neese F (2020) Double-hybrid density functional theory for g-tensor calculations using gauge including atomic orbitals. J Chem Phys 153:054105
Su X-D, Zhang H, Terwilliger TC, Liljas A, Xiao J, Dong Y (2015) Protein crystallography from the perspective of technology developments. Crystallogr Rev 21:122–153
Barth A (2007) Infrared spectroscopy of proteins. Biochim Biophys Acta Bioenerg 1767:1073–1101
Todorovic S, Teixeira M (2018) Resonance Raman spectroscopy of Fe–S proteins and their redox properties. J Biol Inorg Chem 23:647–661
Guo Y, Wang H, Xiao Y, Vogt S, Thauer RK, Shima S, Volkers PI, Rauchfuss TB, Pelmenschikov V, Case DA, Alp EE, Sturhahn W, Yoda Y, Cramer SP (2008) Characterization of the Fe site in iron−sulfur cluster-free hydrogenase (Hmd) and of a model compound via nuclear resonance vibrational spectroscopy (NRVS). Inorg Chem 47:3969–3977
Ogata H, Krämer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, Yoda Y, Tanaka Y, Lubitz W, Cramer SP (2015) Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat Commun 6:7890
Reijerse EJ, Pham CC, Pelmenschikov V, Gilbert-Wilson R, Adamska-Venkatesh A, Siebel JF, Gee LB, Yoda Y, Tamasaku K, Lubitz W, Rauchfuss TB, Cramer SP (2017) Direct observation of an iron-bound terminal hydride in [FeFe]-hydrogenase by nuclear resonance vibrational spectroscopy. J Am Chem Soc 139:4306–4309
Stripp ST (2021) In situ infrared spectroscopy for the analysis of gas-processing metalloenzymes. ACS Catal 11:7845–7862
Pierik AJ, Hulstein M, Hagen WR, Albracht SPJ (1998) A low-spin iron with CN and CO as intrinsic ligands forms the core of the active site in [Fe]-hydrogenases. Eur J Biochem 258:572–578
De Lacey AL, Stadler C, Cavazza C, Hatchikian EC, Fernandez VM (2000) FTIR characterization of the active site of the Fe-hydrogenase from Desulfovibrio desulfuricans. J Am Chem Soc 122:11232–11233
Chen Z, Lemon BJ, Huang S, Swartz DJ, Peters JW, Bagley KA (2002) Infrared studies of the CO-inhibited form of the Fe-Only Hydrogenase from Clostridium pasteurianum I: examination of Its light sensitivity at cryogenic temperatures. Biochemistry 41:2036–2043
Kamp C, Silakov A, Winkler M, Reijerse EJ, Lubitz W, Happe T (2008) Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae. Biochim Biophys Acta 1777:410–416
Ezzaher S, Capon J-F, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Pichon R, Kervarec N, Pe Y, Schollhammer P, Talarmin J, Pichon R, Kervarec N (2007) Evidence for the formation of terminal hydrides by protonation of an asymmetric iron hydrogenase active site mimic. Inorg Chem 46:3426–3428
Barton BE, Rauchfuss TB (2008) Terminal hydride in [FeFe]-hydrogenase model has lower potential for H2 production than the isomeric bridging hydride. Inorg Chem 47:2261–2263
Singleton ML, Bhuvanesh N, Reibenspies JH, Darensbourg MY (2008) Synthetic support of de novo design: sterically bulky [FeFe]-hydrogenase models. Angew Chemie Int Ed 47:9492–9495
Adamska-Venkatesh A, Silakov A, Lambertz C, Rüdiger O, Happe T, Reijerse E, Lubitz W (2012) Identification and characterization of the “super-reduced” state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle? Angew Chemie Int Ed 51:11458–11462
Katz S, Noth J, Horch M, Shafaat HS, Happe T, Hildebrandt P, Zebger I (2016) Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem Sci 7:6746–6752
Chernev P, Lambertz C, Brünje A, Leidel N, Sigfridsson KGV, Kositzki R, Chung-Hung Hsieh SY, Rafael Schiwon MD, Limberg C, Happe T, Haumann M (2014) Hydride binding to the active site of [FeFe]-hydrogenase. Inorg Chem 53:12164–12177
Mebs S, Senger M, Duan J, Wittkamp F, Apfel U-P, Happe T, Winkler M, Stripp ST, Haumann M (2017) Bridging hydride at reduced H-cluster species in [FeFe]-hydrogenases revealed by infrared spectroscopy, isotope editing, and quantum chemistry. J Am Chem Soc 139:12157–12160
Chongdar N, Birrell JA, Pawlak K, Sommer C, Reijerse EJ, Rüdiger O, Lubitz W, Ogata H (2018) Unique spectroscopic properties of the H-cluster in a putative sensory [FeFe] hydrogenase. J Am Chem Soc. https://doi.org/10.1021/jacs.7b11287. (in press)
Land H, Sekretaryova AN, Huang P, Redman HJ, Németh B, Polidori N, Mészáros L, Senger M, Stripp ST, Berggren G (2020) Characterization of a putative sensory [FeFe]-hydrogenase provides new insight into the role of the active site architecture. Chem Sci 11:12789–12801
Birrell JA, Pelmenschikov V, Mishra N, Wang H, Yoda Y, Tamasaku K, Rauchfuss TB, Cramer SP, Lubitz W, DeBeer S (2020) Spectroscopic and computational evidence that [FeFe] hydrogenases operate exclusively with CO-bridged intermediates. J Am Chem Soc 142:222–232
Sommer C, Adamska-Venkatesh A, Pawlak K, Birrell JA, Rüdiger O, Reijerse EJ, Lubitz W (2017) Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J Am Chem Soc 139:1440–1443
Winkler M, Senger M, Duan J, Esselborn J, Wittkamp F, Hofmann E, Apfel U-P, Stripp ST, Happe T (2017) Accumulating the hydride state in the catalytic cycle of [FeFe]-hydrogenases. Nat Commun 8:16115
Rumpel S, Sommer C, Reijerse E, Farès C, Lubitz W (2018) Direct detection of the terminal hydride intermediate in [FeFe] hydrogenase by NMR spectroscopy. J Am Chem Soc 140:3863–3866
Senger M, Laun K, Wittkamp F, Duan J, Haumann M, Happe T, Winkler M, Apfel U-P, Stripp ST (2017) Protonengekoppelte reduktion des katalytischen [4Fe-4S]-Zentrums in [FeFe]-hydrogenasen. Angew Chemie 129:16728–16732
Laun K, Baranova I, Duan J, Kertess L, Wittkamp F, Apfel U-P, Happe T, Senger M, Stripp ST (2021) Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase. Dalt Trans 50:3641–3650
Senger M, Duan J, Pavliuk MV, Apfel U, Haumann M, Stripp ST (2022) Trapping an oxidized and protonated intermediate of the [FeFe]-hydrogenase cofactor under mildly reducing conditions. Inorg Chem 61:10036–10042
Greene BL, Schut GJ, Adams MWW, Dyer RB (2017) Pre-steady-state kinetics of catalytic intermediates of an [FeFe]-hydrogenase. ACS Catal 7:2145–2150
Sanchez MLK, Sommer C, Reijerse E, Birrell JA, Lubitz W, Dyer RB (2019) Investigating the kinetic competency of Cr HydA1 [FeFe] hydrogenase intermediate states via time-resolved infrared spectroscopy. J Am Chem Soc 141:16064–16070
Sanchez MLK, Konecny SE, Narehood SM, Reijerse EJ, Lubitz W, Birrell JA, Dyer RB (2020) The laser-induced potential jump: a method for rapid electron injection into oxidoreductase enzymes. J Phys Chem B 124:8750–8760
Hunt NT (2009) 2D-IR spectroscopy: ultrafast insights into biomolecule structure and function. Chem Soc Rev 38:1837
Senger M, Mebs S, Duan J, Wittkamp F, Apfel U-P, Heberle J, Haumann M, Stripp ST (2016) Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics. Proc Natl Acad Sci USA 113:8454–8459
Horch M, Schoknecht J, Wrathall SLD, Greetham GM, Lenz O, Hunt NT (2019) Understanding the structure and dynamics of hydrogenases by ultrafast and two-dimensional infrared spectroscopy. Chem Sci 10:8981–8989
Kulka-Peschke CJ, Schulz A-C, Lorent C, Rippers Y, Wahlefeld S, Preissler J, Schulz C, Wiemann C, Bernitzky CCM, Karafoulidi-Retsou C, Wrathall SLD, Procacci B, Matsuura H, Greetham GM, Teutloff C, Lauterbach L, Higuchi Y, Ishii M, Hunt NT, Lenz O, Zebger I, Horch M (2022) Reversible glutamate coordination to high-valent nickel protects the active site of a [NiFe] hydrogenase from oxygen. J Am Chem Soc 144:17022–17032
Wrathall SLD, Procacci B, Horch M, Saxton E, Furlan C, Walton J, Rippers Y, Blaza JN, Greetham GM, Towrie M, Parker AW, Lynam J, Parkin A, Hunt NT (2022) Ultrafast 2D-IR spectroscopy of [NiFe] hydrogenase from E. coli reveals the role of the protein scaffold in controlling the active site environment. Phys Chem Chem Phys 24:24767–24783
Tai H, Nishikawa K, Higuchi Y, Mao Z-WW, Hirota S (2019) Cysteine SH and glutamate COOH contributions to [NiFe] hydrogenase proton transfer revealed by highly sensitive FTIR spectroscopy. Angew Chemie Int Ed 58:13285–13290
Ash PA, Carr SB, Reeve HA, Skorupskaite A, Rowbotham JS, Shutt R, Frogley MD, Evans RM, Cinque G, Armstrong FA, Vincent KA (2017) Generating single metalloprotein crystals in well-defined redox states: Electrochemical control combined with infrared imaging of a NiFe hydrogenase crystal. Chem Commun 53:5858–5861
Ash PA, Kendall-Price SET, Evans RM, Carr SB, Brasnett AR, Morra S, Rowbotham JS, Hidalgo R, Healy AJ, Cinque G, Frogley MD, Armstrong FA, Vincent KA (2021) The crystalline state as a dynamic system: IR microspectroscopy under electrochemical control for a [NiFe] hydrogenase. Chem Sci 12:12959–12970
Acknowledgements
Sven T. Stripp is funded by the German Research Foundation (DFG) within the framework of SPP 1927 priority program “Iron-Sulfur for Life” (Grant No. STR1554/5-1). Jason W. Sidabras is funded by the Medical College of Wisconsin, while some results published in this work had funding from the European Union Horizon 2020 Marie Skłodowska-Curie Fellowship (no. 745702; ACT-EPR, https://act-epr.org) and the Max Planck Society.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sidabras, J.W., Stripp, S.T. A personal account on 25 years of scientific literature on [FeFe]-hydrogenase. J Biol Inorg Chem 28, 355–378 (2023). https://doi.org/10.1007/s00775-023-01992-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-023-01992-5