
Abstract
Glycogen phosphorylase (GP) is biologically active as a dimer of identical subunits, each activated by phosphorylation of the serine-14 residue. GP exists in three interconvertible forms, namely GPa (di-phosphorylated form), GPab (mono-phosphorylated form), and GPb (non-phosphorylated form); however, information on GPab remains scarce. Given the prevailing view that the two GP subunits collaboratively determine their catalytic characteristics, it is essential to conduct GPab characterization to gain a comprehensive understanding of glycogenolysis regulation. Thus, in the present study, we prepared rabbit muscle GPab from GPb, using phosphorylase kinase as the catalyst, and identified it using a nonradioactive phosphate-affinity gel electrophoresis method. Compared with the half-half GPa/GPb mixture, the as-prepared GPab showed a unique AMP-binding affinity. To further investigate the intersubunit communication in GP, its catalytic site was probed using pyridylaminated-maltohexaose (a maltooligosaccharide-based substrate comprising the essential dextrin structure for GP; abbreviated as PA-0) and a series of specifically modified PA-0 derivatives (substrate analogs lacking part of the essential dextrin structure). By comparing the initial reaction rates toward the PA-0 derivative (Vderivative) and PA-0 (VPA-0), we demonstrated that the Vderivative/VPA-0 ratio for GPab was significantly different from that for the half-half GPa/GPb mixture. This result indicates that the interaction between the two GP subunits significantly influences substrate recognition at the catalytic sites, thereby providing GPab its unique substrate recognition profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycogen, a highly branched polymer of d-glucose (G), serves as a form of energy storage in animals, fungi, and bacteria (Roach et al. 2012; Prats et al. 2018; Katz 2022). A key enzyme for utilizing glycogen is glycogen phosphorylase (GP; EC 2.4.1.1; MW 1.9 × 105), which catalyzes the sequential phosphorolysis of glycogen to release α-d-glucose 1-phosphate (G-1-P) (Titani et al. 1977; Tagaya and Fukui 1984; Roach et al. 2012; Prats et al. 2018; Katz 2022). GP is biologically active as a dimer of identical subunits (Dombrádi 1981), and each subunit is activated by phosphorylase kinase (PhK; EC 2.7.11.19) through phosphorylation of the serine-14 (Ser14) residue (Krebs et al. 1964; Dombrádi 1981; Chan and Graves 1982; Nadeau et al. 2018). There are thus three isolatable GP forms, namely GPa (di-phosphorylated form, high activity), GPab (mono-phosphorylated form, moderate activity), and GPb (non-phosphorylated form, low activity) (Dombrádi 1981). Muscle GP activity is regulated by interconversion between these three forms and the binding of various allosteric effectors, such as AMP, ATP, and d-glucose 6-phosphate (G-6-P) (Madsen et al. 1983). The most highly activated forms of muscle GP are AMP bound, regardless of the phosphorylation state (Lowry et al. 1964; Rush and Spriet 2001).
GPa and GPb have been well characterized by structural (Madsen et al. 1983) and kinetic (Lowry et al. 1964; Madsen et al. 1983; Rush and Spriet 2001) approaches, including X-ray crystallography (Sprang et al. 1988, 1991; Barford et al. 1991). In contrast, little research progress has been achieved on GPab, because the preparation and identification of the phospho–dephospho hybrid is highly challenging. Nevertheless, there is a prevailing view that the two GP subunits collaboratively determine their catalytic characteristics (Fig. 1), as supported by numerous experimental results (Burkhardt and Wegener 1994; Buchbinder et al. 1995; Rath et al. 2000; Mathieu et al. 2017; Kish et al. 2023). Therefore, GPab characterization is indispensable for a comprehensive understanding of glycogenolysis regulation. Notably, Burkhardt and Wegener (1994) reported that the AMP-binding affinity of hawk moth muscle GPab (Kd, ~ 30 μM) was significantly different from those of both GPa (Kd, ~ 0.3 μM) and GPb (Kd, ~ 300 μM), suggesting that phosphorylation of either subunit of the GP dimer partially, but not fully, changed the AMP-binding site structures of both subunits. Accordingly, we are interested in whether the intersubunit communication in GP similarly influences substrate recognition at the catalytic sites, thereby providing GPab with a unique substrate recognition profile.

Prevailing view about phosphorylation regulation of GP. The two GP subunits collaboratively determine their catalytic characteristics, and therefore, the catalytic characteristics of GPab (mono-phosphorylated form) are essentially different from those of GPa (di-phosphorylated form) and GPb (non-phosphorylated form) (Burkhardt and Wegener 1994)
GP assay is usually performed using macromolecular glycogen (MW 106–107) as the substrate. Notably, here, each GP subunit has two distinct maltooligosaccharide binding sites, namely a storage and catalytic site (Barford et al. 1991). Because the affinity for maltooligosaccharide is approximately 20 times higher in the storage site (Kd, ~ 1 mM) than in the catalytic site (Kd, ~ 20 mM), the GP dimer binds macromolecular glycogen mainly through the two storage sites to form a GP–glycogen complex (Makino et al. 2015). Conformational changes in one or both GP subunits may affect the positional relationship between the two storage sites, which might result in an affinity change of GP for macromolecular glycogen. However, quantitative evaluation of this effect on the GP activity is very difficult. Therefore, to focus on the catalytic site activity, small maltooligosaccharides comprising the minimum essential dextrin structure for GP should be used as the assay substrate (Makino et al. 2015). From this viewpoint, we previously developed pyridylaminated-maltohexaose (G-G-G-G-G-F, F = 1-deoxy-1-[(2-pyridyl)amino]-d-glucitol]; abbreviated as PA-0) and a series of specifically modified PA-0 derivatives (Gm-Z-Gn-F, m + n = 4 and Z = 3-acetoamido-3-deoxy-d-altrose) to investigate substrate recognition at the GP catalytic site (Nakamura et al. 2017). In the present study, by using PA-0 and its derivatives, the substrate recognition profile of GPab was compared with those of GPb, GPa, and a half-half GPa/GPb mixture. In addition, their substrate recognitions were compared by using branched dextrins such as G-G-G-(G↔)G-G-G-F and G-G-G-G-(G↔)G-G-F (the double-headed arrow represents an α-1,6-glycosidic bond).
Materials and methods
Materials
Rabbit muscle glycogen phosphorylase a (GPa), glycogen phosphorylase b (GPb), phosphorylase kinase (PhK), AMP, ADP, and ATP were purchased from Sigma-Aldrich (St. Louis, MO, USA). The TSKgel DEAE-5PW column (7.5 × 75 mm) was acquired from Tosoh (Tokyo, Japan) and the Shodex NH2P-50 column (4.6 × 150 mm) was purchased from Showa Denko (Tokyo, Japan). The Vivaspin 6 centrifugal concentrator [3.0 × 104 molecular weight cut-off (MWCO)] was acquired from Sartorius Stedim Lab (Gloucestershire, UK). Sodium dodecyl sulfate (SDS), WIDE-VIEW prestained protein size marker III, SuperSep Ace 7.5% polyacrylamide gel, and SuperSep Phos-tag (50 μmol/L) 6% polyacrylamide gel were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). Coomassie brilliant blue (CBB; trade name EzStainAQua) was acquired from ATTO (Tokyo, Japan).
GPa and GPb were further purified according to previously described methods (Burkhardt and Wegener 1994). The fluorogenic oligosaccharides Gn-F (n = 0–5) and Gm-Z-Gn-F (m + n = 4) were prepared using our previously reported methods (Nakamura et al. 2017). The fluorogenic branched dextrins G-G-(G↔)G-G-G-G-F (BD-3), G-G-G-(G↔)G-G-G-F (BD-4), G-G-G-G-(G↔)G-G-F (BD-5), and G-G-G-G-G-(G↔)G-F (BD-6) were also prepared using our previously reported methods (Yamamoto et al. 2007).
Preparation of rabbit muscle GPab
A mixture (300 μL) containing 50 mM Tris–HCl buffer (pH 8.2), 3.0 mM ATP, 10 mM MgCl2, 0.30 mM CaCl2, 40 mM sodium fluoride, 3.0 mM 2-mercaptoethanol, 0.30 mg rabbit muscle GPb, and 2.0 U rabbit muscle PhK was incubated at 37 °C for 10 min (Miyagawa et al. 2016). One unit of PhK was defined as the amount of enzyme that produces 1.0 μg of GPa per minute under the employed conditions. Next, the product solution was immediately subjected to anion-exchange HPLC using a TSKgel DEAE-5PW column (7.5 × 75 mm) at 25 °C and a flow rate of 0.9 mL/min. Two eluents, A and B, were employed. Eluent A comprised 5.0 mM sodium 2-glycerophosphate buffer, pH 6.8, containing 150 mM d-glucose and Eluent B comprised 30 mM sodium 2-glycerophosphate buffer, pH 6.8, containing 150 mM d-glucose and 500 mM NaCl. The column was first equilibrated with Eluent A. After injecting the sample, linear gradient elutions were performed using the following Eluent A:Eluent B proportions (v/v) and times: 100:0 in 5 min, 50:50 in 35 min, 0:100 in 5 min, 0:100 in 5 min, and 100:0 in 1 min. Each elution was monitored by measuring the absorbance at 280 nm. An intermediate protein exhibiting significant GP activity was collected as Fraction X. This fraction was then concentrated to 300 μL using a Vivaspin 6 centrifugal concentrator (3.0 × 104 MWCO) and washed five times with a 3.0 mL solution containing 40 mM sodium phosphate buffer (pH 6.8) and 1.0 mM 2-mercaptoethanol. The enzyme solution was stored at 4 °C and used within 48 h.
Polyacrylamide gel electrophoresis (PAGE)
Normal SDS-PAGE was performed after reduction of the sample with 2-mercaptoethanol, using a SuperSep Ace 7.5% polyacrylamide gel, as described by Laemmli (1970). WIDE-VIEW prestained protein size marker III was used as the MW marker. After electrophoresis, proteins in the gel were stained with CBB using EzStainAQua.
Improved Phos-tag SDS-PAGE (Zn2+–Phos-tag SDS-PAGE) was performed after reduction of the sample with 2-mercaptoethanol, using a SuperSep Phos-tag (50 μmol/L) 6% polyacrylamide gel (FUJIFILM Wako Pure Chemical), as described by Kinoshita and Kinoshita-Kikuta (2011). Following electrophoresis, the proteins in the gel were stained with CBB using EzStainAQua.
GP assay in the direction of glycogenolysis
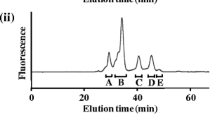
GP assay was performed in the direction of glycogenolysis according to our previously reported method (Nakamura et al. 2017). Briefly, a mixture (70 μL) containing 40 mM sodium phosphate buffer (pH 6.8), 35 μM PA-oligosaccharide, 1.0 mM 2-mercaptoethanol, 0.05% gelatin, 0–10 mM AMP, and 5–500 nM GP (i.e., 0.01–1 μM GP subunit) was incubated at 37 °C for 30 min. To terminate the reaction, the mixture was heated at 100 °C for 5 min. The chain-shortened product was then isolated and quantified by size-fractionation HPLC using a Shodex NH2P-50 (4.6 × 150 mm) column at a flow rate of 0.9 mL/min and 25 °C (Nakamura et al. 2017). The eluent, comprising acetonitrile:water:acetic acid (750:250:3, v/v/v), was titrated to pH 7.0 with 5.0% aqueous ammonia. PA-oligosaccharides were detected by fluorescence (excitation wavelength, 320 nm; emission wavelength, 400 nm). Three independent experiments were performed for each condition.
Although the fluorogenic substrate concentration was very low, it was considered to be constant throughout the reaction period because the chain-shortened product could be measured even at 10 fmol and > 95% of the original substrate remained unchanged at the end of the reaction period.
Results and discussion
Schematic representation of the GP catalytic site
A schematic representation of the GP catalytic site is shown in Fig. 2a. The GP catalytic site is composed of six subsites (Si, Sii, Siii, Siv, Sv, and Sp) that are complementary to five tandem d-glucose residues (Gi, Gii, Giii, Giv, and Gv) and one phosphate (Pi) molecule, respectively (Weber et al. 1978; Johnson et al. 1988; Nakamura et al. 2017). In the direction of glycogenolysis, the α-1,4-glycosidic linkage between Gi and Gii is split, forming G-1-P. Although the catalytic action of GP is essentially reversible, a high concentration of Pi in the body inhibits GP activity in the direction of glycogen synthesis in animals. Previously, we reported that the apparent Km and Vmax values toward PA-0 were similar to those toward methyl α-maltopentaoside (G5-OCH3) (Nakamura et al. 2017). This indicates that the nonreducing end maltopentaosyl (G5-) residue of PA-0 suitably fits all the Si–Sv subsites.

Probing of the GP catalytic site using fluorogenic oligosaccharides. a Schematic representation of the productive binding of PA-0 and Pi to the GP catalytic site. The GP catalytic site is composed of six subsites (Si, Sii, Siii, Siv, Sv, and Sp), wherein Si and Sp are the sites for the nonreducing end G residue and Pi, respectively (Weber et al. 1978; Johnson et al. 1988; Nakamura et al. 2017). When Gi, Gii, Giii, Giv, and Gv of the maltooligosaccharide substrate and Pi interact with Si, Sii, Siii, Siv, Sv, and Sp, respectively, the α-1,4-glycosidic linkage between Gi and Gii is phosphorolyzed. b Structures and designations of the fluorogenic oligosaccharides used in this study. G = d-glucose residue; Z = 3-acetoamido-3-deoxy-d-altrose residue; F = 1-deoxy-1-[(2-pyridyl)amino]-d-glucitol residue; hyphens represent α-1,4-glycosidic bonds; and the double-headed arrow represents an α-1,6-glycosidic bond
Preparation and identification of rabbit muscle GPab
GP was the first enzyme studied in detail in relation to phosphorylation regulation (Krebs et al. 1964), with many GP studies performed using rabbit muscle GPa and/or GPb (Lowry et al. 1964; Madsen et.al. 1983; Sprang et al. 1988, 1991; Barford et al. 1991; Rush and Spriet 2001). Notably, while rabbit muscle GPa, GPb, and PhK are commercially available, GPab is not. This is one of the major reasons for the stagnation of GPab research. Previously, some dedicated researchers prepared GPab in vitro by the partial phosphorylation of GPb using PhK as the catalyst (Vereb et al. 1987, 1992; Harris and Graves 1990). Notably, in the PhK-catalyzed GPb phosphorylation reaction, production of GPa (di-phosphorylated form, secondary product) is inevitable even at an early stage of the reaction, because the phosphorylation rate toward GPab (mono-phosphorylated form, primary product) is faster than that toward GPb (non-phosphorylated form, original substrate) (Harris and Graves 1990). In addition, to correctly perform spectrophotometric protein detection (absorbance at 280 nm, A280), GP proteins should be completely separated from ATP and ADP, which have strong UV light-absorbing properties and are abundant in the product solution.
To perform the GPab study correctly and consistently, we modified previously reported procedures for the preparation and identification of GPab: First, PhK-catalyzed phosphorylation of rabbit muscle GPb was performed according to our previously reported method (Miyagawa et al. 2016), except that gelatin (protein adsorption-preventing agent) and ethylenediaminetetraacetic acid (EDTA; PhK inactivator) were not added to prevent disruption of the subsequent HPLC purification. Instead, the reaction product was immediately subjected to DEAE-5PW anion-exchange HPLC. The HPLC conditions were similar to those previously reported by Harris and Graves (1990), except that the initial concentration of sodium 2-glycerophosphate buffer was reduced from 30 to 5 mM; however, its concentration gradually increased (5–17.5 mM) in parallel with the NaCl gradient elution (0–250 mM). Notably, glycerophosphate ions in the mobile phase competitively inhibited four phosphorous components (i.e., GPab, GPa, ADP, and ATP) to bind to the DEAE-5PW resins. Through our modifications, these four phosphorous components were separated from each other (Fig. 3), whereas separation and detection of ADP was not considered in the previous study (Harris and Graves 1990). An intermediate protein exhibiting significant GP activity was collected as Fraction X. Although some previous studies adopted an instantaneous increase in the eluent glycerophosphate concentration [e.g., jumping from 30 to 200 mM (Vereb et al. 1987, 1992)] to eluate GPab from the anion-exchange column, we found that this practice caused co-elution of GPab and GPa (data not shown).

DEAE-5PW anion-exchange high-performance liquid chromatogram of the partial phosphorylation product of GPb. Rabbit muscle GPb was partially phosphorylated in vitro by rabbit muscle PhK, and the reaction product was immediately subjected to DEAE-5PW anion-exchange HPLC. a Product from 10 min reaction in the absence of ATP; dotted lines represent the gradient elution pattern. The small peak at 5 min was attributed to the presence of a contaminant. b Product from 10 min reaction in the presence of 3 mM ATP. Open circles indicate the relative GP activity in the eluted solution. The fraction indicated by the black bar was collected as Fraction X. c Product from 120 min reaction in the presence of 3 mM ATP
GPab has been usually identified through a radioactive labeling method using [γ-32P]ATP as the substate (Vereb et al. 1987, 1992; Harris and Graves 1990). However, the use of radioactive materials significantly hinders routine performance in GPab research because researchers must follow strict rules to order, store, use, and dispose of these materials. To avoid this, in the present study, GPab was identified by a new nonradioactive method using normal (Phos-tag-free) and Phos-tag SDS-PAGEs (Kinoshita and Kinoshita-Kikuta 2011). Phos-tag, a dinuclear metal complex of 1,3-bis[bis(pyridin-2-ylmethyl)amino]propan-2-olato, is a phosphate-binding molecule, and has been applied to nonradioactive analyses of protein phosphorylation reactions (Kinoshita et al. 2006). In Phos-tag SDS-PAGE, Phos-tag residues covalently linked to polyacrylamide gel interfere with phosphorylated protein migration, resulting in the separation of the phosphorylated form, as a retarded band, from the non-phosphorylated form. The results of both SDS-PAGEs are shown in Fig. 4. For the normal SDS-PAGE, Fraction X migrated as a single band of MW 9.7 × 104, which was concordant with the MW of a GP subunit. However, with Phos-tag SDS-PAGE, Fraction X split into two bands corresponding to the phosphorylated and non-phosphorylated GP subunits. From these results, Fraction X was identified as the phospho–dephospho hybrid of GP, specifically, GPab.

Identification of Fraction X by normal and Phos-tag SDS-PAGEs. GPa, GPb, and Fraction X were also analyzed using normal and Phos-tag SDS-PAGEs. The proteins in the gel were stained with CBB. a Normal SDS-PAGE: Fraction X migrated as a single band of MW 9.7 × 104, which is concordant with the MW of a GP subunit. b Phos-tag SDS-PAGE: Phos-tag residues covalently linked to polyacrylamide gel interfered with the migration of the phosphorylated protein, resulting in the separation of the phosphorylated form, as a retarded band, from the non-phosphorylated form (Kinoshita and Kinoshita-Kikuta 2011). Fraction X split into two bands corresponding to the phosphorylated and non-phosphorylated GP subunits
AMP-induced activation of rabbit muscle GPab
Muscle GP is allosterically activated by AMP (Lowry et al. 1964; Madsen et al. 1983; Rush and Spriet 2001). Notably, muscle GPa exhibits a very strong affinity for AMP (Kd, ~ 0.3 μM) compared with muscle GPb (Kd, ~ 300 μM) (Miyagawa et al. 2016). As previously stated, Burkhardt and Wegener (1994) reported that GPab from hawk moth muscle showed a single and unique AMP dependence (Kd, ~ 30 μM), suggesting that phosphorylation of either subunit of the GP dimer partially, but not fully, changed the AMP-binding site structures of both subunits. As for rabbit muscle GPab, the AMP dependence was previously compared with that of GPb (Vereb et al. 1987); however, comparison of the three phosphorylation forms (i.e., GPa, GPab, and GPb) is necessary to fully characterize GPab. In addition, comparison of GPab with a half-half GPa/GPb mixture is expected to provide clear criteria for assessing the interaction between the phosphorylated and non-phosphorylated subunits (Fig. 1). Hence, in the present study, we compared the AMP dependence in rabbit muscle GPa, GPab, GPb, and a half-half GPa/GPb mixture. To focus on the catalytic site activity, PA-0, comprising the minimum essential dextrin structure for GP, was used as the assay substrate (Fig. 2); in contrast, macromolecular glycogen was used in the previous GPab studies (Vereb et al. 1987, 1992; Harris and Graves 1990; Burkhardt and Wegener 1994). The results are summarized in Fig. 5: The AMP-induced activation profile of rabbit muscle GPab showed a single-step rise curve, which fell between those of rabbit muscle GPa and GPb. This agrees with the results of the study by Burkhardt and Wegener (1994) on insect muscle GPab activity toward macromolecular glycogen. In addition, the AMP-induced activation profile of the half-half GPa/GPb mixture presented a two-step rise curve, which was remarkably different from that of GPab. These results indicated that the two heterogenous subunits of rabbit muscle GPab significantly interacted with each other (Fig. 1), inducing the unique AMP-binding affinity of both subunits.

Effect of AMP on the catalytic activity of rabbit muscle GPs with various phosphorylation states. The initial reaction rates toward PA-0 at various AMP concentrations for GPa, GPb, and GPab were compared. Each series is expressed as a percentage of the respective maximum activity. A common logarithmic scale is used for the abscissa to show a very wide ATP concentration range. Each value shows the mean ± SD (n = 3)
Substrate recognition at the GPab catalytic sites
The maltooligosaccharide-binding region of the GP catalytic site accommodates and recognizes the nonreducing end maltopentaosyl (G5-) residue of a substrate, as shown in Fig. 2a (Nakamura et al. 2017). By comparing the initial reaction rate toward the PA-0 derivative (Vderivative) with that toward PA-0 (VPA-0), we previously reported that Vderivative/VPA-0 decreased with increasing GP activation level (Nakamura et al. 2017). Specifically, as the GP activation level increases, GP catalytic site recognition of the G5 residue must become stricter. In the present study, using PA-0 (G5-F) and its derivatives (Gm-Z-Gn-F, m + n = 4) as substrates, the substrate recognition profile of GPab was compared with those of GPa, GPb, and the half-half GPa/GPb mixture. The Vderivative/VPA-0 ratio for GPab fell between the GPa and GPb ratios, as expected (Table 1 and Fig. 6). Remarkably, the Vderivative/VPA-0 ratio for GPab was much greater than that for the half-half GPa/GPb mixture, suggesting that the phosphorylated subunit of GPab was not sufficiently activated compared with that of GPa. This suggestion was supported by the lower activity of GPab compared to that of the half-half GPa/GPb mixture (Footnotes d and e in Table 1).

Effect of the AltNAc residue position on the GP activity. Using GPs at various activation levels, the initial reaction rates toward the PA-0 derivatives were compared with that toward PA-0. Chemical structures of PA-0 and its derivatives are shown in Fig. 2b. A natural logarithmic scale is used for the ordinate to show a wide range of the Vderivative/VPA-0 ratios. Data for AMP-activated GPb, GPa, and GPb were reproduced from our previous report (Nakamura et al. 2017). Each value shows the mean ± SD (n = 3). Our previous study revealed that with increasing GP activation level, recognition of the substrate maltopentaosyl (G-G-G-G-G-) residue by the GP catalytic site became increasingly stricter
We next proceeded to compare the substrate recognitions of GPa, GPab, GPb, and the half-half GPa/GPb mixture using branched dextrins G-G-G-(G↔)G-G-G-F (BD-4), G-G-G-G-(G↔)G-G-F (BD-5), and G-G-G-G-G-(G↔)G-F (BD-6). The results are summarized in Table 2 and Fig. 7. The VBD-6/VPA-0 ratios approximated unity regardless of the GP activation level, indicating that the α-1,6-linked G residue of BD-6 was located outside of the GP catalytic site. Interestingly, the VBD-5/VPA-0 ratios were significantly lower than the VPA-5/VPA-0 ratios. This might be because the α-1,6-linked G residue is bulky, and thus, in addition to the Sv–Gv interaction, the Siv–Giv interaction might also be somewhat disturbed (Fig. 2a). Similar to the VPA-4/VPA-0 and VPA-5/VPA-0 ratios (Table 1 and Fig. 6), the VBD-4/VPA-0 and VBD-5/VPA-0 ratios for GPab were significantly greater than those for the half-half GPa/GPb mixture (Table 2 and Fig. 7).

Effect of the α-1,6-linked G residue position on the GP activity. Using GPs at various activation levels, the initial reaction rates toward BD isomers were compared with that toward PA-0. Chemical structures of PA-0 and BD isomers are shown in Fig. 2b. A natural logarithmic scale is used for the ordinate to show a wide range of the VBD isomer/VPA-0 ratios. Each value shows the mean ± SD (n = 3)
As we expected, the three phosphorylation forms of GP displayed the respective substrate recognition profiles. Notably, substrate recognition of GPab was significantly different from that of the half-half GPa/GPb mixture. These results indicated that the intersubunit communication in GP significantly influenced substrate recognition at the catalytic sites. A deeper structural investigation (e.g., via X-ray crystallography or hydrogen–deuterium-exchange mass spectrometry) would be helpful to obtain further information.
Notably, because GP catalyzes the first step in glycogenolysis, it has become a potential key target for treating type 2 diabetes (Oikonomakos and Somsák 2008). Hence, the search for potent inhibitors of the GP catalytic site is attracting particular attention. Based on the in vitro inhibitory effects toward GPa and AMP-activated GPb, several d-glucose analogs, such as β-d-glucopyranosylamines, C-β-d-glucopyranosyl derivatives, and iminosugars, were reported as hopeful candidates (Somsák 2011). These d-glucose analogs predominantly bind to the Si subsite (Fig. 2a) and competitively inhibit a maltooligosaccharide substrate from binding to the catalytic site. In the present study, it was revealed that GPab catalytic sites have unique catalytic characteristics due to the interaction between the phosphorylated and non-phosphorylated subunits. Accordingly, we are planning to investigate the inhibitory effects of these d-glucose analogs toward GPab, which may provide important information toward their practical use in the medical field.
Availability of data and material
The data that supported the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Barford D, Hu SH, Johnson LN (1991) Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J Mol Biol 218:233–260
Buchbinder JL, Guinovart JJ, Fletterick RJ (1995) Mutations in paired α-helices at the subunit interface of glycogen phosphorylase alter homotropic and heterotropic cooperativity. Biochemistry 34:6423–6432
Burkhardt G, Wegener G (1994) Glycogen phosphorylase from flight muscle of the hawk moth, Manduca sexta: purification and properties of three interconvertible forms and the effect of flight on their interconversion. J Comp Physiol B 164:261–271
Chan KFJ, Graves DJ (1982) Rabbit skeletal muscle phosphorylase kinase. J Biol Chem 257:5948–5955
Dombrádi D (1981) Structural aspects of the catalytic and regulatory function of glycogen phosphorylase. Int J Biochem 13:125–139
Harris WR, Graves DJ (1990) Kinetic analysis of the separate phosphorylation events in the phosphorylase kinase reaction. Arch Biochem Biophys 276:102–108
Johnson LN, Cheetham J, McLaughlin PJ, Acharya KR, Barford D, Phillips DC (1988) Protein–oligosaccharide interactions: lysozyme, phosphorylase, amylases. Curr Top Microbiol Immunol 139:81–134
Katz A (2022) A century of exercise physiology: key concepts in regulation of glycogen metabolism in skeletal muscle. Eur J Appl Physiol 122:1751–1772
Kinoshita E, Kinoshita-Kikuta E (2011) Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 11:319–323
Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteom 5:749–757
Kish M, Subramanian S, Smith V, Lethbridge N, Cole L, Vollmer F, Bond NJ, Phillips JJ (2023) Allosteric regulation of glycogen phosphorylase by order/disorder transition of the 250’ and 280s loops. Biochemistry 62:1360–1368
Krebs EG, Love DS, Bratvold GE, Trayser KA, Meyer WL, Fischer EH (1964) Purification and properties of rabbit skeletal muscle phosphorylase b kinase. Biochemistry 3:1022–1033
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lowry OH, Schult DW, Passonneau JV (1964) Effects of adenylic acid on the kinetics of muscle phosphorylase a. J Biol Chem 239:1947–1953
Madsen NB, Shechosky S, Fletterick RJ (1983) Site-site interactions in glycogen phosphorylase b probed by ligands specific for each site. Biochemistry 22:4460–4465
Makino Y, Fujii Y, Taniguchi M (2015) Properties and functions of the storage sites of glycogen phosphorylase. J Biochem 157:451–458
Mathieu C, Dupret JM, Lima FR (2017) The structure of brain glycogen phosphorylase–from allosteric regulation mechanisms to clinical perspectives. FEBS J 284:546–554
Miyagawa D, Makino Y, Sato M (2016) Sensitive, nonradioactive assay of phosphorylase kinase through measurement of enhanced phosphorylase activity towards fluorogenic dextrin. J Biochem 159:239–246
Nadeau OW, Fontes JD, Carlson GM (2018) The regulation of glycogenolysis in the brain. J Biol Chem 293:7099–7107
Nakamura M, Makino Y, Takagi C, Yamagaki T, Sato M (2017) Probing the catalytic site of rabbit muscle glycogen phosphorylase using a series of specifically modified maltohexaose derivatives. Glycoconj J 34:563–574
Oikonomakos NG, Somsák L (2008) Advances in glycogen phosphorylase inhibitor design. Curr Opin Investig Drugs 9:379–395
Prats C, Graham TE, Shearer J (2018) The dynamic life of the glycogen granule. J Biol Chem 293:7089–7098
Rath VL, Ammirati M, LeMotte PK, Fennell KF, Mansour MN, Danley DE, Hynes TR, Schulte GK, Wasilko DJ, Pandit J (2000) Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol Cell 6:139–148
Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS (2012) Glycogen and its metabolism: some new developments and old themes. Biochem J 441:763–787
Rush JWE, Spriet LL (2001) Skeletal muscle glycogen phosphorylase a kinetics: effects of adenine nucleotides and caffeine. J Appl Physiol 91:2071–2078
Somsák L (2011) Glucose derived inhibitors of glycogen phosphorylase. C R Chimie 14:211–223
Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterick RJ, Madsen NB, Johnson LN (1988) Structural changes in glycogen phosphorylase induced by phosphorylation. Nature 336:215–221
Sprang SR, Withers SG, Goldsmith EJ, Fletterick RJ, Madsen NB (1991) Structural basis for the activation of glycogen phosphorylase b by adenosine monophosphate. Science 254:1367–1371
Tagaya M, Fukui T (1984) Catalytic reaction of glycogen phosphorylase reconstituted with a coenzyme-substrate conjugate. J Biol Chem 259:4860–4865
Titani K, Koide A, Hermann J, Ericsson LH, Kumar S, Wade RD, Walsh KA, Neurath H, Fisher EH (1977) Complete amino acid sequence of rabbit muscle glycogen phosphorylase. Proc Natl Acad Sci USA 74:4762–4766
Vereb G, Fodor A, Bot G (1987) Kinetic characterization of rabbit muscle phosphorylase ab hybrid. Biochim Biophys Acta 915:19–27
Vereb G, Pallagi E, Gergely P (1992) Phosphorylation-induced conformational changes in the phosphorylase ab hybrid as revealed by resolution of pyridoxal 5’-phosphate with imidazole citrate cysteine. Mol Cell Biochem 110:113–121
Weber IT, Johnson LN, Wilson KS, Yeates DG, Wild DL, Jenkins JA (1978) Crystallographic studies on the activity of glycogen phosphorylase b. Nature 274:433–437
Yamamoto E, Makino Y, Omichi K (2007) Active site mapping of amylo-α-1,6-glucosidase in porcine liver glycogen debranching enzyme using fluorogenic 6-O-α-glucosyl-maltooligosaccharides. J Biochem 141:627–634
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. NK, AI, and YM: performed the experiments, analyzed the data, and wrote the first draft of the manuscript. YM and HM: revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethics approval
This work does not include any studies with human participants or animals.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Handling editor: F. Polticelli.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kamada, N., Ikeda, A., Makino, Y. et al. Intersubunit communication in glycogen phosphorylase influences substrate recognition at the catalytic sites. Amino Acids 56, 14 (2024). https://doi.org/10.1007/s00726-023-03362-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00726-023-03362-6