
Abstract
The combination of molecularly imprinted polymers (MIPs) and inverse opals (IO) have been a point of interest in the past few years due to their potential in sensing applications. At the same time, peptide nucleic acid (PNA) is a stable analogue to natural occurring genetic material. In this study, we describe the preparation and characterization of a PNA imprinted matrix, based on the controlled self-assembly of organized silica particles (SiPs) arrays. The degree of organization of the silica arrays are compared to the organization of the cavities after the removal of the SiPs, using spatial statistical analysis. This analysis of the Voronoi tessellations, pair correlation functions and bond order showed that the successfully formed arrays contain a high degree of quasi-hexagonal (hexatic) organization of the cavities, with both global and local order. The adsorption analysis of the materials show potential for developing future materials with tunable structural reflective properties, such as on-site, color- changing genetic material sensor.
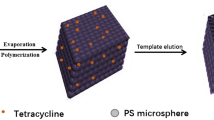
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent years, peptide nucleic acids (PNAs) have been increasingly receiving attention due to their resemblance to naturally occurring nucleic acids such as deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) [1]. In contrast to DNAs with a negatively charged sugar-phosphate backbone, PNAs have a neutral pseudopeptide backbone consisting of N-(2-aminoethyl)glycine units (Fig. 1a) which results in a lack of electrostatic repulsion between a synthetic PNA and a DNA backbone.

Peptide Nucleic Acid. a PNA general structure and b antisense property between an exemplary PNA strand containing all four nucleobases and a complementary DNA
In addition, the similar distances between the nucleobases within the particular strands lead to a high affinity and specificity of PNAs towards Watson–Crick complementary DNA strands (Fig. 1b) [2]. PNAs can, therefore, perfectly act as structural DNA or RNA mimic [3]. These unique properties of PNAs further result in a higher chemical and enzymatic stability and fewer side effects, which is why PNAs have found various applications in biomedicine, such as antisense therapy, gene-detection or drug development [4]. One of these potential applications is as a template molecule for polymer imprinting, which can be used as a biosensor of genetic material.
Molecularly imprinted polymers (MIPs) are commonly explained as being synthetic receptors for a targeted molecule, being often described as analogues to natural antibody–antigen systems. MIPs (the antibodies) are fabricated with specific recognition sites towards the template molecule (the antigen). During the synthesis step the template molecules and functional monomers are self-assembled in a solvent that will later serve as porogen. During the polymerization a crosslinking agent is used to fix the recognition sites. The self-assembly occurs when the template molecule and the functional monomers interact with each other, and after the polymerization the imprinting site is fixed. The template is later removed, and the specific recognition cavity is left behind in the polymeric matrix, which now can selectively attract the target molecule. The general process of molecular imprinting is shown in Fig. 2.

Process of molecular imprinting. The template molecule is mixed with the functional monomers and binds with them through non-covalent or covalent interactions. The formed pre-polymer solution contains functional monomers bound to the template. The polymer is then polymerized with a crosslinking agent, and afterwards, the template molecule is removed leaving behind specific recognition cavities with functional groups. To achieve recognition of the target molecule, the cavities bind specifically the molecule of interest
This technology is often compared to a “lock and key” mechanism [5]. In 1973, Wulff reported molecularly imprinted polymers for the first time [6], and since then this technology has attracted widespread attention. MIPs are smart adsorption materials attractive for their highly specific recognition ability for a variety of targets, and they have shown superior chemical and physical stability, in contrast to biological receptors. Some of the application fields are: solid phase extraction, immunoassays, chromatographic separation, depollution, sensing, cell imaging, chiral recognition, and biomedicine [7,8,9,10,11,12,13,14,15,16,17].
More recently, MIPs are being used in combination with inverse opals as a base for photonic crystals (PhC) for a signaling output of the sensing properties of MIPs [18,19,20]. PhCs can be described as periodic arrays formed by homogenously shaped spheres or layers with specific optical properties in a host material (usually a polymer). To enable a photonic band gap (PBG), the materials involved need to have different refractive indexes, thus creating a difference in dielectric constants [21, 22]. One of these materials could be air, in which case either the organized arrays are self-assembled, or the host material is highly porous. This description is also applied to the well-known opal and inverse opal (IO) structures [23,24,25]. For an inverse opal to function as a photonic crystal, it needs to have a high level of organization, since defects in the lattice lead to unwanted photon propagation [26, 27]. The reflected wavelength from the photonic crystals is highly dependent on two factors: the distance between the layers or sphere, and the refractive index. If one of these variables is changed, for example, by changing the thickness of the layers or the diameter of the spheres via a bio-chemical sensor, or by the attachment of a third component on the surface of the layers, the reflected wavelength also changes [26]. Because of their characteristics, PhCs based on IO have become the ideal sensing output for MIPs, in contrast to traditional biosensors. The combination of these two technologies provides a platform to actively adjust its structural color in a specific environment through the design of its chemical components. Molecularly imprinted PhCs based on IO sensors are showing potential in the fields of rapid screening and point-of-care diagnostics with label-free, visual, and on-site detection [14, 28,29,30,31,32,33,34]. For instance, studies show the ability of MIP PhCs to detect harmful components in food, such as histamine [35]. Because the quality of IO is so highly dependent on the homogeneity of particles and layers that conform it, the spheres’ polydispersity index (PDI) and degree of organization is of outmost importance. Less than 0.05 is considered monodisperse [36, 37]; however, the degree of organization is not usually quantifiable and in most cases it is described by the observation of scanning electron microscope (SEM) or transmission electron microscope (TEM).
In this study, we present a method of fabricating PNA imprinted polymeric inverse opal, and use spatial statistical analysis of the pore locations, using the disLocate package [38] to examine the degree of organization.
Results and discussion
To begin with, the PNA template molecule was synthesized via liquid phase synthesis. The PNA was characterized via high-performance liquid chromatography (HPLC) and electrospray ionization mass spectrometry (ESI–MS), respectively. Then we synthesized highly homogeneous silica particles following our previous work [39]. The SiPs were characterized via SEM and DLS, where the diameter and the hydrodynamic diameter (Dh) were obtained. The opalescent arrays were self-assembled via evaporation deposition. This material was characterized by scanning electron microscopy (SEM) and the organization of the lattice was analyzed using spatial statistical analysis. A pre-polymer solution containing the PNA template molecule was infiltrated thought the lattice, this system was then polymerized. To obtain the imprinted recognition cavities, the silica particles and the template were removed using hydrofluoric acid. The final inverse opal was also analyzed using SEM and spatial statistics, to describe quantitatively the degree of organization, comparing the inverse opal to the original silica opal.
Synthesis of PNA
In contrast to typically applied solid phase PNA synthesis, an improved liquid phase synthesis strategy is used to synthesize the PNA template [1]. This is a more efficient and ecological method further enabling the application of standard techniques of synthetic organic chemistry as well as the facile monitoring of reaction processes. A protected PNA monomer is deprotected and coupled to a hydrophobic anchoring group in an apolar solvent, allowing the intermediates and the product to precipitate by addition of a polar solvent. This method enables an easy purification by simply washing the precipitate. Further monomers can be added likewise. In the case of the used PNA dimer, two adenine monomers were added. The free backbone amine of the desired PNA was capped with acetic anhydride to avoid side reactions and deprotection, and cleavage of the anchoring group finally lead to the desired PNA template (see Fig. 3). The formation was verified by detecting the exact mass of the molecule with ESI–MS and the purity was measured by using HPLC.

Synthesis of PNA dimer. Compound 1 and adenine PNA monomer 2 were dissolved in dichloromethane (DCM), 1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholinocarbenium hexafluorophosphate (COMU) and N,N-diisopropylethylamine (DIPEA) were added to the solution. After the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and piperidine, the mixture was stirred for 5 min and subsequently neutralized with 1 N HCl. The solvent was removed under reduced pressure and the residue was precipitated in MeOH, then it was washed several times with MeOH and acetonitrile and dried to obtain compound 3. These steps were repeated to obtain compound 4 which was then dissolved in DCM. Pyridine and acetic anhydride were added and the mixture was stirred for 3 h. The solvent was removed under reduced pressure and the residue was precipitated in MeOH, washed several times with MeOH and acetonitrile and dried to obtain compound 5 which was then added to a mixture of trifluoroacetic acid (TFA): triisopropylsilane (TIPS): water and stirred for 3 h. The volatiles were removed under reduced pressure and the residue was precipitated in iPr2O. After drying, the desired PNA dimer 6 was obtained as a white solid with an overall yield of 73%
Synthesis of silica particles (SiPs) and formation of organized arrays
The synthesis of the SiPs is based on the Stöber method [40]. It took place in two steps, the hydrolysis of TEOS to form silanol monomers, and then condensation to give way to a siloxane network, which allowed the formation of nuclei and subsequent growth. The reaction was stirred for 3 h and then stopped by centrifugation followed by consequent washing. Precision and consistency in this step are the key factors to control the size of the synthesized SiPs while keeping a low PDI.
The average SiPs diameter size was measured via SEM, and the average hydrodynamic diameter was measured via DLS. For significant validation of the results obtained from the SEM, 50 different points were measured of 5 different samples of SiPs for each experiment. In case of DLS, 5 aliquots were measured from 15 different samples. The average particle diameter used was 150 ± 3.1 nm, and a hydrodynamic average diameter (Dh X̅) of 178.8 ± 0.6 nm was determined. The difference in the two diameter values was expected since the latter one refers to moving particles in a dispersion. These results are shown in Fig. 4a, b, respectively. To obtain high quality and organized arrays only samples with PDI lower than 0.05 were used.

Characterization of the SiPs opals and inverse opals. a SEM image of the synthesized SiPs. Measuring diameters directly from the SEM yields an average diameter of 150 ± 3.1 nm. b Hydrodynamic sizes of the SiPs were measured via DLS. The average DLS result shows an average hydrodynamic diameter of 178.8 ± 0.6 nm. The hydrodynamic diameter is usually a higher value than the SEM-based diameter since it comes from moving particles in a solution. c SEM observation of the SiP photonic crystals. The sample used for these figures were made using 1% of suspended particles in 12 cm3 of EtOH via solvent evaporation method. The inserts in this image show the correlation map or fast Fourier transform. d The non-imprinted polymeric (NIP) matrix, obtained after the removal of the silica particles via HF treatment. e The molecularly imprinted polymeric (MIP) matrix. The sample was polymerized with a solution containing the template molecule. Later, the silica particles and the template were removed with an HF treatment. Both the NIP and MIP appear to have a high degree of organization, but somewhat lower than the original silica particles opal arrays. However, the MIP shows holes on the surface (circled in red), which probably originate from the template conglomerates. f Cross-section of the molecularly imprinted matrix. (All SEM scale bars correspond to 1 µm)
The opalescent organized arrays were formed using 1% w/w of SiPs in ethanol on a piranha solution treated glass slide (2 × 2 cm) via evaporation deposition method. SEM was used to characterize the top view of the formed PhC. These results show the high homogeneous morphology and the high degree of organization. Visually, it is possible to recognize the hexagonal pattern formed by the SiPs (shown in red on Fig. 4c). This is a characteristic trait of the face-centered-cubic (FCC) and hexagonal-closed-packed (HCP) arrangement. And it is also supported by the coordination cloud or fast Fourier transform (FFT) of the image, insert in the top right-hand corner (Fig. 4c), showing six distinct equally spaced lobes as expected for good hexagonal close packing.
Formation of inverse opal
During the formation of the inverse opal, two parallel procedures have been followed as follows: one using a pre-polymer solution that contains the template molecules to obtain the MIP; and a second sample that follows the same conditions but without containing the template molecule; the resulting material is the non-imprinted polymer (NIP). This is a control sample to evaluate the specific adsorption capacity of the MIP. The pre-polymer solution was prepared as described in the experimental method, using methacrylic acid (MAA) as functional monomer and ethylene glycol dimethacrylate (EGDMA) as crosslinker. The combination was chosen due to its non-covalent interaction with PNA (Scheme 1).

The organized silica particles were sandwiched between the glass slide where they were assembled and a poly(methyl methacrylate) (PMMA) slide. Using a syringe, the degassed pre-polymer solution was infiltrated between the slides and the voids between the SiPs. This step had to be performed with extreme care, since the ethanol-based pre-polymer solution could wash out the particles and disrupt their organization. Then the sandwiched system was photo-polymerized for 4 h with a 365 nm UV light. After this step, the samples were submerged in 1% hydrofluoric acid (HF). This solution actively attacks silica, thus separating the glass slide from the PMMA and etching the particles. In case of the MIP, the PNA template molecule is also removed with this step. Because of the low concentration of HF, the samples had to be stirred in this solution for 72 h, refreshing it every 24 h.
The resulting material was observed using scanning electron microscopy, and the NIP is shown in Fig. 4d, next to the organized silica particles, for the ease of comparison. The MIP inverse opal can be seen in Fig. 4e. Both the MIP and NIP inverse opals retained a high organization, supported by their FFT of the image (insert in the top right hand corner of Fig. 4d, e), showing six distinct globes. However, even by naked eye it is possible to determine that honey-comb-like arrays have been disrupted, most likely during the infiltration process. The MIPs lattice also contains bigger holes, circled in red in the figure. These might be caused by agglomeration of silica particles, or impurities of the polymer solution. A cross-section of the PNA molecularly imprinted material is shown in Fig. 4f. Some defects on the lattice can also be seen. The high porosity of the matrix makes it unsuitable for spatial characterization analysis.
Characterization of the molecularly imprinted inverse opal
The analysis of the adsorption properties of the molecularly imprinted inverse opal was done by re-binding experiments using the template PNA as analyte. The adsorption equilibrium was obtained after shaking the samples in PNA containing solutions at 20 °C. It was determined that they reach a maximum binding adsorption after 72 h. However, a difference between the imprinted inverse opal (MIP-IO) and the control non-imprinted inverse opal (NIP-IO) can be observed from the first hour (Fig. 5a) of the shaking procedure.

PNA MIP-IO and NIP-IO adsorption analysis. a Adsorption kinetic curve. The equilibrium was determined based on the results of the samples shaken with 1 mM solution. b Adsorption isotherm curves. c Langmuir and Freundlich isotherm models fittings to the adsorption curves. All the adsorption experiments were carried out at 20 °C
The rebinding effect of the imprinted inverse opal increased not only with time but also with concentration of PNA. To determine the binding capacity of the PNA MIP-IO, saturation adsorption experiments were performed at different initial concentration. The solutions prepared were of 0.1, 0.2, 0.5, 0.8, 1.0, 2.0, and 5.0 mM, and the binding capacities thus achieved were 1.330, 5.104, 7.507, 12.968, 16.042, 31.241, and 35.952 µmol/g of MIP-IO, respectively. The NIP also showed an increase of the binding capacity to 0.511, 2.292, 4.111, 6.228, 6.590, 7.952, 11.354 µmol/g of NIP-IO. However, the NIP has a clearly inferior capacity in terms of specific binding compared to the imprinted material, the MIP-IO. According to Fig. 5b, the amount of PNA bound to the inverse opal increased rapidly when the concentration was below 1 mM, but it seems to reach a plateau at 5 mM. The maximum adsorption capacities of the MIP-IO and NIP-IO were 35.952 µmol/g and 11.354 µmol/g, respectively; this clear difference is an indication of the formation of specific recognition cavities due to the imprinting process.
The reusability of the imprinted material was tested by repeating the experiment 5 times with the same concentration of PNA in the solution (1 mM). The intermediate washing was carried out with the same conditions used to initially remove the silica particles and the template molecule. The imprinted material was found to be very stable and maintain an adsorption capacity at an average of 91.5%. Table 1 shows the results of this test.
Adsorption isotherms
To further understand the binding behavior of the material, the experimental data was analyzed with the Freundlich (Eq. 1) and Langmuir isotherm model (Eq. 2) as follows:
where Qm (μmol/g) is the apparent maximum adsorbed amount, Ce (μmol/cm3) is the equilibrium concentration, b (g/cm3) is the adsorption equilibrium constant of Langmuir, Kf (μmol/g) is an indicative constant for the adsorption capacity of the sorbent, and the constant 1/n indicates the intensity of the adsorption. A value of n higher than 1 indicates a favorable adsorption of the molecules onto the adsorbent surface. A value of higher n reflects the higher degree of adsorption [41].
The Freundlich model describes the empirical relationship between the quantity of the concentration of a solute that is adsorbed onto the surface of a solid and the concentration of the solute in the liquid phase. On the other hand, the Langmuir model assumes that ions are adsorbed as a monolayer on the surface. The Freundlich equation can also be written as shown in Eq. (3), which fits the description of a linear equation, whilst the Langmuir equation can be viewed as a hyperbola as follows:
Figure 5c shows the Langmuir and Freundlich isotherm models for PNA adsorption onto MIP-IO and NIP-IO. The figure shows that the Langmuir isotherm model fitted the experimental data significantly better than that of the Freundlich isotherm model. The adsorption isotherm parameters are listed in Table 2. The values of the correlation coefficients for the PNA MIP-IO (R2 = 0.94642) and NIP-IO (R2 = 0.97847) by the Langmuir isotherm model were higher than those calculated by the Freundlich isotherm model, which indicates that the produced materials explained by the Langmuir isotherm model. These results were expected since the Langmuir models better describes a chemical interaction between the adsorbate and the surface, instead of the physical interaction described by the Freundlich model. A better fitting of the Langmuir models also supports the expected result of a monolayer of adsorbates (in our case PNA) in the imprint cavities capable of reaching a maximum equilibrium, in contrast to the multilayer that the Freundlich model describes.
Spatial analysis of the molecularly imprinted inverse opal
To describe quantitatively the structure and assign a value to the relative order, the formed inverse opals were characterized from SEM micrographs with spatial statistics obtained using the Mathematica package, disLocate (Detecting Intermolecular Structure Located at particle positions) [38].
Local-scale order can be divided into the following three distinct categories: translational, entropic, and angular. Translational order is present when each particle within the system occupies a precise position that repeats at a fixed distance, determined by a specific translation period. Entropic order is attained when the system has the minimal amount of unoccupied space, maximizing its density. Angular order refers to the arrangement of the "bonds" or contact points between a particle and its neighboring particles, specifically in terms of their relative arc-separation [42,43,44]. When the neighbors of a given particle exhibit identical angular arc symmetry and equidistant spacing, and the covering area is maximized due to the equivalent positioning of all particles in relation to each other, it represents complete periodicity. In order to evaluate the various types of order in silica particle arrays and inverse opals, different spatial order metrics were utilized on the images. Local free volume and the complementary metric, covering area [45] can be calculated by partitioning the substrate into a Voronoi tessellation around each individual particle [46]. To determine the boundaries of each cell in the tessellation, a line perpendicular to the midpoint is drawn along the line-of-sight vector that connects the nearest neighbors surrounding each particle. The intersection points of these lines establish the unit cell that encloses each particle. The area covered by these cells is then extracted and compared to the expected coverage for a hexagonally close packed system, revealing the distribution of hexagonally packed regions. Additionally, a coordination number can be defined by examining the number of facets in the Voronoi cells that contain the particle, akin to chemical valence states [47,48,49]. This coordination number is derived from the entropic driving force, which drives the alignment of faceted or functionalized particles to maximize system entropy by minimizing free volume (refer to the inset in Fig. 4c–e for further details).
In order to illustrate different local order states, the Voronoi tessellation maps can be color-coded based on several factors. These factors include the deviation of the cell area from the expected value for ideal hexagonal packing, the coordination number, or the deviation of the bond order parameter from the expected symmetry value (which will be further explained below). By applying these color codes, the variations in local order can be visually highlighted.
Going beyond the first neighbor, it is possible to quantify the positional order probabilistically using the pair correlation function [50]. The pair correlation function, g(r), quantifies the number of neighboring objects within a small shell at a distance r from a central particle, averaged over all particles in the system. By visualizing the distances between neighbors as a set of Voronoi cells emanating from each particle, the collective sum of these cells provides an estimation of the average configuration (r). In a system with a periodic lattice, the objects are uniformly distributed at specific distances through repeated translations of a unit cell. The level of order can be determined by examining the number of peaks in the pair correlation function, with characteristic spacing between the peaks corresponding to each specific Bravais lattice.
Angular orientation order describes the likelihood of finding an object at a given angular arc-separation between neighboring particles, most commonly thought of as the symmetry state of a system. Angular order can be calculated by using the bond order parameter [51], which compares the angle between the central particle and its closest neighbors against a specified symmetry basis vectors (Fig. S4c, f). In the aggregate across all particles, this can be collapsed down into a coordination cloud or fast Fourier transform (FFT) of the image [52, 53]. In systems where circles are densely packed, hexagonal configurations offer the highest density, making the sixfold bond order values (q6) particularly significant. These methods have been extensively employed to assess disorder, identify regions with polycrystalline or disordered characteristics, and determine the likelihood of intermolecular spacings [54, 55]. The comprehensive spatial statistical analysis for each PhC array formed is summarized in Fig. S1-S3.
The spatial characterization of the silica particles show a high degree of order, as discussed previously [39] with nearest neighbor spacing slightly less than the measured particle diameters from SEM. There are also visible cracks and defects in the periodic arrangement; however, the surface is highly organized, with regions between the cracks showing a high degree of local order. Nearly 40% of particles correspond to pure hexagonal packing [39] as shown by the Voronoi tessellations, colored by the deviation from a perfect lattice (see supporting info Fig. S1). Almost 90% of particles have six nearest neighbors, supporting a quasi-hexatic lattice formation. After templating the inverse opals, both with and without the molecular imprinting, the ordering is disrupted. However, the films still contain a high degree of local order. Interestingly, the MIP films contain roughly 60% of the number of pores observed in the NIP films. The pores in the MIP film are on average larger (148 ± 24 nm vs. 114 ± 15 nm for MIP and NIP films respectively) and more widely spaced (207 ± 2.2 nm vs. 157 ± 1.8 nm, respectively) Although the inverse opals both showed decreased ordering relative to the Si particles, they still retained a quasi-hexatic structure. The MIP was slightly more disturbed showing nearly 30% fewer sixfold neighbors relative to the original SiP opals, compared to only 23% for the NIP film, and a greater lattice disorder parameter [38]. However, the local order between the defects were quite similar for both films having 68% of particles in a quasi-hexatic structure, with less than 10% deviation in the Voronoi areas from that of a perfect hexagonal lattice (see supporting Fig. S2 and S3, c and d). Although there was some observed agglomeration with molecular imprinting, there was little influence on the local ordering of the resultant inverse opal films.
Conclusion
In the present study a unique PNA molecularly imprinted inverse opal polymeric array was synthesized. A PNA dimer was used as template molecule for the imprinting process. The highly organized silica particles arrays give way to also organized inverse opals, however the spatial characterization of the arrays showed that the degree of organization decreases possibly because of the pre-polymer infiltration process. In future investigations as an alternative to the Mathematica DisLocate software, the spatial characterization could also be performed using the Characteristic Crystallographic Element (CCE) norm [56]. The polymerization procedure and the subsequent treatment with hydrofluoric acid created a highly porous material with non-covalent recognition sites. The adsorption experiments for the PNA showed that imprinted material had a much higher binding capacity compared to the non-imprinted one. The maximum adsorption capacities of the MIP-IO and NIP-IO were 35.952 µmol/g and 11.354 µmol/g, respectively. The nearly three times higher adsorption is an indication of formation of specific recognition cavities during the imprinting process. The reusability experiment showed that molecularly imprinted inverse opal could be reused at least five times without reducing their adsorption capacities. Due to the high degree of organization of the MIP-IO this material has the potential for a rapid photonic structural coloration change, leading to future on-site genetic material recognition devices, such as the rapid identification of bacteria, or viruses that are health endangering.
Experimental
Tetraethyl orthosilicate (TEOS, 98%), and ammonia (NH3, 28% w/w) from Alfa Aesar (Karlsruhe, Germany). Ethanol (EtOH, ABS) used for the different stages of the synthesis and work-up was purchased from ChemLab (Zedelgem, Belgium). The water (18 MΩ cm pure water) was treated with a Milli-Q Gradient system, Millipore Corp, to keep conditions constant. For the glass slides treatment, a 1:4 piranha solution (H2O2/H2SO4) was used. Hydrogen peroxide (33.3%) was purchased from VWR Chemicals and sulphuric acid (95–97.0%) was received from Merck. Ethylene glycol dimethacrylate (EGDMA, 98.0%), azobisisobutyronitrile (AIBN), and methacrylic acid (MAA, 99.0%) were purchased from Sigma-Aldrich. Hydrofluoric acid (1%) was obtained from MicroChemicals. N,N-Diisopropylethylamine was purchased from Carl Roth, distilled over KOH, decanted, refluxed over NaH, distilled and stored under nitrogen atmosphere. Triisopropylsilane (TIPS, 99.0%) and piperidine (99.0%) were purchased from Sigma Aldrich. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 98.0%) and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU, 98.0%) were purchased from Fluorochem and trifluoroacetic acid (TFA, 99.0%) was purchased from Alfa Aesar or Fluorochem respectively. Diisopropyl ether (iPr2O) was purchased from Merck. All reagents were used as received unless otherwise stated.
Characterization methods
Dynamic light scattering (DLS) (Zetasizer Nano, Malvern Panalytical GmbH) measurements were carried out to determine the average hydrodynamic diameter (Dh) and the polydispersity index (PDI). In this study, the PDI refers to the uniformity of the particles, (where a value close to 0.0 is a perfect homogeneity of the sample, while a 1.0 is a sample with a large variety of sizes). The samples were prepared in 0.1 M solution with Milli-Q water as dispersant in disposable cuvettes (DTS 0012), sonicated for 30 min. The DLS measurements via intensity were performed at 25 °C without filtration before the measurements. SEM images were obtained with a Jeol 6400 (Jeol, Peabody, MA, USA, with an operating voltage EHT = 5 kV). The optical observation of the SiPs was carried out with an optical microscope at 200 × magnification (ZEISS axio imager A1m—Carl Zeiss AG). The reflection spectra were measured using a spectrometer (LAMBDA 1050 + UV/VIS/NIR Spectrometer with 150 mm InGaAs Int. Sphere and UV WinLab software—PerkinElmer Inc.). To characterize the organized silica arrays and photonic crystals, the same coating conditions and SEM were used. LC–MS analysis of the PNA dimer was done on an Agilent Series 1100 HPLC system (column: Kinetex 2.6 u, C18, 100A, 50 × 4.6 mm) with an Agilent 6520 QTOF with electrospray ionization (ESI) in the positive mode using a gradient elution with 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile (eluent B) from 0 to 1 min 97:3, to 11 min 87:13, to 21 min 57:43 to 23 min 0:100, to 26 min 0:100 to 30 min 97:3. To test the adsorption properties of the molecularly imprinted cavities, the MIP and NIP samples were shaken for 48 h in different concentrations of PNA solutions with a shaking water bath 1086 (GFL—Gesellschaft für Labortechnik mbH). The rebinding capacity of the fabricated matrix was analyzed with an HPLC (Agilent Infinity 1290 RRLC—Rapid Resolution Liquid Chromatography (column: Kinetex 2.6 u, C18, 100A, 50 × 4.6 mm)—Agilent Technologies Inc.). The samples for this analysis were first purified with a 0.22-µm pore filter (Millipak® Express 40). Samples were eluted via gradient elution using eluent A (0.1% TFA in water) and eluent B (0.1% TFA in acetonitrile) from 0 to 1 min 97:3, to 4.3 min 87:13, to 6 min 57:43, to 6.6 min 10:90, to 7 min 10:90 to 8 min 97:3. A UV Vis diode array detector at 260 nm was used for detection. The degree of organization was determined with spatial statistical analysis, using the Mathematica package ‘disLocate’, a suite of tools to rapidly quantify the spatial structure of a two-dimensional dispersion of objects. The name ‘disLocate’ stands for “detecting intermolecular structure located at particle positions” [38].
Synthesis of PNA
The synthesis procedure is shown in Fig. 3. Compound 1 (0.80 mmol) and the adenine PNA monomer 2 (1.04 mmol) were dissolved in dichloromethane (DCM) (80 cm3), 1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholinocarbenium hexafluorophosphate (COMU, 1.04 mmol) and N,N-diisopropylethylamine (DIPEA, 4.0 mmol) were added and the solution was stirred for 10 min. After the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 10.7 mmol) and piperidine (16.2 mmol), the mixture was stirred for 5 min and subsequently neutralized with 1 N HCl (28.8 mmol). The solvent was removed under reduced pressure and the residue was precipitated in MeOH, washed several times with MeOH and acetonitrile and dried to obtain compound 3. These steps were repeated to obtain compound 4 which was then dissolved in DCM (80 cm3). Pyridine (49.6 mmol) and acetic anhydride (12.8 mmol) were added and the mixture was stirred for 3 h. The solvent was removed under reduced pressure and the residue was precipitated in MeOH, washed several times with MeOH and acetonitrile and dried to obtain compound 5 (Fig. 3) which was then added to a mixture of trifluoroacetic acid (TFA): triisopropylsilane (TIPS): water (95:2.5:2.5, 60 cm3) and stirred for 3 h. The volatiles were removed under reduced pressure and the residue was precipitated in diisopropyl ether (iPr2O). After drying, the desired PNA dimer 6 (0.58 mmol) was obtained as a white solid with an overall yield of 73%. The product was analyzed via LC–MS. ESI–MS: m/z calculated for C26H36N16O6 [M + 2H]2+ 334.1497, found 334.1511.
Synthesis of SiPs and self-assembly
The silica particles (SiPs) were synthesized following the Stöber method [40], through the controlled hydrolysis and condensation of tetraethyl orthosilicate (TEOS) in alcohol medium. Based on the previous synthesis and characterization of SiPs [38], we synthesized SiPs of 150 nm diameter in average. To achieve this, 100 cm3 of ethanol were mixed with 8 cm3 of ammonia (80 mmol) and 3 cm3 of water in a 250 cm3 round bottom flask at 60 °C for 10 min. 6 cm3 of TEOS (26 mmol) were added slowly with the addition pump set to 18 cm3/h. Constant and controlled addition speed allows reproducibility of the particles size. After the full addition of all reagents, the reaction mixture was stirred for 3 h and then stopped by centrifugation to prevent further nucleation. It was then washed three times with 50 cm3 of EtOH to remove all unreacted monomers.
The organizing self-assembly of the particles was performed via the evaporative deposition method using a 1% suspension of SiPs in 12 cm3 of EtOH. The glass slides where the opalescent arrays of silica are sedimented were first treated with a 1:4 piranha solution (H2O2/H2SO4) for 1 h, with the purpose of increasing their hydrophilicity. The glass slides were then rinsed with several aliquots of Milli-Q water and dried with a stream of nitrogen.
The glass slides were then inserted vertically in conical-bottom centrifuge tubes. Figure 6a exemplifies the formation of the opalescent films on the piranha solution treated glass. After the ethanol was evaporated completely, the SiPs organized array was obtained on both sides of the glass slides, which appeared as an opalescent structure. To prepare this opal structure for the infiltration of the pre-polymer solution, it was covered with a clean poly(methyl-methacrylate) (PMMA) 2 × 2 cm slide and held together using clamps on each side. An exemplification of the infiltration process is shown in Fig. 6b.

Fabrication methodology for the formation of SiPs based opals and inverse opals. a To obtain the organized arrays of SiPs, the particles were fully suspended in 1% w/w in ethanol, within a conical bottom centrifuge tube. A piranha solution treated glass slide was carefully placed vertically in the tube. The system is placed in a quiet, vibration free environment, where the ethanol evaporates at constant rate. The suspended particles self-assemble on the glass due to capillary forces in an organized matter as the evaporation takes places. b For the infiltration of the pre-polymer solutions, the formed SiP opals are sandwiched between the glass slide on which they were deposited and a clean polymethyl-methacrylate (PMMA) slide. The system was held together with clamps on each side. Using a syringe, the pre-polymer solution was infiltrated through the system and the voids between the silica particles
Infiltration of pre-polymer solution
This solution contained the functional monomer (methacrylic acid, MAA), the PNA template molecule, the solvent (EtOH) that serves as porogen and the crosslinker (ethylene glycol dimethacrylate, EGDMA). 0.4 cm3 of MAA (4.718 mmol) were mixed with 0.2 cm3 EGDMA (1.060 mmol) and 1 mg of AIBN as initiator in 0.4 cm3 ethanol (6.85 mmol). To this solution, the template molecule was added to the crosslinker in a ratio of 1:6 that is 118 mg of the synthesized PNA (0.177 mmol). The mixture was degassed with nitrogen for 20 min. The degassed pre-polymer solution was infiltrated through the opening between the glass and the PMMA slide holding the SiPs opal. This system was then photo-polymerized in a UV chamber at 365 nm for 4 h. To realize the imprinted cavities, the glass-SiP/polymer-PMMA sandwiched system was immersed in 1 wt% HF aqueous solution at room temperature for 72 h to remove the silica nanoparticles and the template molecule. A schematic showing the formation of the imprinted inverse opal is shown in Fig. 6b. Scheme 1 shows the formation of the imprinted array and demonstrated the rebinding ability of the material.
Adsorption experiments of the PNA MIP-IO
MilliQ water was used as the solvent, for all adsorption experiments. PNA solutions of different concentration were prepared (0.1 mM, 0.2 mM, 0.5 mM, 0.8 mM, 1 mM, 2 mM, and 5 mM). The inverse opal with the PMMA slide as support was weighted to determine the mass of the imprinted material, and then it was placed horizontally in a container. 15 cm3 of the prepared PNA solutions were added and then the containers were sealed tightly with paraffin and shaken at 20 °C. To determine the binding equilibrium time, an aliquot of the solution was analyzed via HPLC to determine the concentration at given time. This test was performed at 1, 5, 8, 24, 32, 48, 56, and 72 h after the start of the re-binding procedure. As shown in Fig. 5a, a binding equilibrium starts to show after 50 h, but it is more stable after 72 h. For the HPLC analysis, the solutions were prefiltered through a 0.22-µm membrane. The adsorption capacity (Q) was calculated using Eq. (4). For this study, the average of 5 replicates were used to produce the data. The average weight of the molecularly imprinted inverse opal was determined to be 17.8 ± 0.5 mg. However, individual weight was used to calculate the adsorbing capacity of the IO.
where C0 (mmol/dm3) is the initial PNA concentration molecule, Ct (mmol/dm3) is the PNA concentration of the template molecule at a given time (t). With the exception of the experiments to determine the binding equilibrium, all the re-binding experiments were shaken for 72 h. V (dm3) is the total volume of the solution; m (mg) is mass of the MIP or NIP material.
Data availability
Not applicable.
References
Jancik Prochazkova A, Gaidies S, Yumusak C, Brüggemann O, Weiter M, Sariciftci NS, Scharber MC, Čépe K, Zbořil R, Krajcovic J, Salinas Y, Kovalenko A (2020) Mater Today Chem 17:100272
Siddiquee S, Rovina K, Azriah A (2015) Adv Tech Biol Med 3:131
Shiraishi T, Nielsen PE (2014) Methods Mol Biol 1050:193
Gupta A, Mishra A, Puri N (2017) J Biotechnol 259:148
Brüggemann O, Visnjevski A, Burch R, Patel P (2004) Anal Chim Acta 504:81
Wulff G, Sarhan A, Zabrocki K (1973) Tetrahedron Lett 14:4329
Kubo T, Nomachi M, Nemoto K, Sano T, Hosoya K, Tanaka N, Kaya K (2006) Anal Chim Acta 577:1
Sarafraz-Yazdi A, Razavi N (2015) Trends Anal Chem 73:81
Song YP, Zhang L, Wang GN, Liu JX, Liu J, Wang JP (2017) Food Control 82:233
Lian Z, Li H-B, Wang J (2016) Anal Bioanal Chem 408:5527
Ahmad OS, Bedwell TS, Esen C, Garcia-Cruz A, Piletsky SA (2019) Trends Biotechnol 37:294
Su C, Li Z, Zhang D, Wang Z, Zhou X, Liao L, Xiao X (2020) Biosens Bioelectron 148:111819
Goud KYMS, Reddy KK, Gobi KV (2016) J Chem Sci 128:763
Chen L, Wang X, Lu W, Wu X, Li J (2016) Chem Soc Rev 45:2137
Leibl N, Haupt K, Gonzato C, Duma L (2021) Chemosensors 9:123
Canfarotta F, Rapini R, Piletsky S (2018) Curr Opin Electrochem 7:146
da Fonseca Alves R, da Silva LNC, Matos Neto G, Ierick IF, Ferreira TL, Sotomayor MDPT (2021). In: Martín-Esteban A (ed) Molecularly imprinted polymers methods in molecular biology, vol 2359. Humana, New York
Cui F, Zhou Z, Zhou HS (2020) Sensors 20:996
Herrera-Chacón A, Cetó X, del Valle M (2021) Anal Bioanal Chem 413:6117
Mazzotta E, Di Giulio T, Malitesta C (2022) Anal Bioanal Chem 414:5165
Hou J, Li M, Song Y (2018) Angew Chem Int Ed 57:2544
Sukhoivanov IA, Guryev IV (2009). In: Sukhoivanov IA, Guryev IV (eds) Photonic crystals: physics and practical modeling 152. Springer, Heidelberg, p 1
Armstrong E, O’Dwyer C (2015) J Mater Chem C 3:6109
Yeo SJ, Choi GH, Yoo PJ (2017) J Mater Chem A 5:17111
Abdurakhmonov SD, Ashurov MS, Klimonsky SO, Tcherniega NV (2022) Bull Lebedev Phys Inst 49:137
Fenzl C, Hirsch T, Wolfbeis OS (2014) Angew Chem Int Ed 53:3318
Sukhoivanov IA, Guryev IV (2009). In: Sukhoivanov IA, Guryev IV (eds) Photonic crystals: physics and practical modeling, 159. Springer, Heidelberg, p 67
Chen W, Meng Z, Xue M, Shea KJ (2016) Mol Imprinting 4:1
Chen C, Liu Y, Wang H, Chen G, Wu X, Ren J, Zhang H, Zhao Y (2018) ACS Nano 12:10493
Chen Q, Shi W, Cheng M, Liao S, Zhou J, Wu Z (2018) Microchim Acta 185:557
Han MG, Shin CG, Jeon S-J, Shim H, Heo C-J, Jin H, Kim JW, Lee S (2012) Adv Mater 24:6438
Barahona F, Albero B, Tadeo JL, Martín-Esteban A (2019) J Chromatogr A 1587:42
Barahona F, Díaz-Álvarez M, Turiel E, Martín-Esteban A (2016) J Chromatogr A 1442:12
Pan J, Li L, Hang H, Wu R, Dai X, Shi W, Yan Y (2013) Langmuir 29:8170
Ahmed S, Ansari A, Haidyrah AS, Chaudhary AA, Imran M, Khan A (2022) ACS Appl Polym Mater 4:2783
Danaei M, Dehghankhold M, Ataei S, Hasanzadeh Davarani F, Javanmard R, Dokhani A, Khorasani S, Mozfari MR (2018) Pharmaceutics 10:57
Mudalige T, Qu H, van Haute D, Ansar SM, Paredes A, Ingle T (2019). In: López Rubio A, Fabra Rovira MJ, Martínez Sanz M, Gómez Gómez-Mascaraque L (eds) Micro and nano technologies, nanomaterials for food applications. Elsevier, Berlin
Bumstead M, Liang K, Hanta G, Hui LS, Turak A (2018) Sci Rep 8:1554
Estrada Alvarez SA, Guger I, Febbraro J, Turak A, Lin H-R, Salinas Y, Brüggemann O (2022) Materials 15:5864
Stöber W, Fink A, Bohn E (1968) J Colloid Interface Sci 26:62
Bao L, Meng M, Sun K, Li W, Zhao D, Li H, He M (2014) J Appl Polym Sci 131:40890
Agarwal U, Escobedo FA (2011) Nat Mater 10:230
Timmermans J (1961) J Phys Chem Solids 18:1
Bernard EP, Krauth W (2011) Phys Rev Lett 107:155704
Williamson JJ, Evans RML (2014) J Chem Phys 141:164901
Okabe A, Boots B, Sugihara K, Chiu SN, Kendall DG (2000) Spatial Tessellations. John Wiley & Sons Inc, Hoboken, NJ, USA
van Anders G, Ahmed NK, Smith R, Engel M, Glotzer SC (2014) ACS Nano 8:931
Agarwal U, Escobedo FA (2012) J Chem Phys 137:024905
Damasceno PF, Engel M, Glotzer SC (2012) ACS Nano 6:609
Kirkwood JG, Boggs EM (1942) J Chem Phys 10:394
Steinhardt PJ, Nelson DR, Ronchetti M (1983) Phys Rev B 28:784
Magonov SN, Whangbo M-H (2008) Surface analysis with STM and AFM: Experimental and theoretical aspects of image analysis. VCH, Weinheim
Bharati MH, Liu JJ, MacGregor JF (2004) Chemom Intell Lab Syst 72:57
Rankin DWH, Mitzel NW, Morrison CA (2013) Structural methods in molecular inorganic chemistry. Wiley, Chichester
Ladd M, Palmer R (2013) Structure determination by X-ray crystallography. Springer, New York
Ramos PM, Herranz M, Foteinopoulou K, Karayiannis NC, Laso M (2020) Crystals 10:1008
Acknowledgements
We thank the funding support of the Chemical Monthly Fellowship (MoChem) of the Austrian Academy of Sciences (ÖAW), the Linz Institute of Technology (LIT), Johannes Kepler University Linz, project number LIT413760001 CHAMS, and Natural Science and Engineering Council of Canada, project number RGPIN- 2019-05994, and the Ontario Ministry of Research and Innovation project number ER15-11-123. The open access was provided by Johannes Kepler Open Access Publishing Fund.
Funding
Open access funding provided by Johannes Kepler University Linz.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Estrada, S.A.A., Gaidies, S., Febbraro, J. et al. Spatial characterization of peptide nucleic acid molecularly imprinted inverse opal. Monatsh Chem 154, 925–938 (2023). https://doi.org/10.1007/s00706-023-03091-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-023-03091-2