
Abstract
Several 1-benzyl and 1,3-dibenzyl derivatives of tetrahydropyridinylidene salts with differing electron withdrawing substituents at the aromatic residues have been prepared. In addition, the amine moiety in position 4 was varied. The new compounds were investigated for their antiplasmodial and antitrypanosomal activities as well as for their cytotoxicity. They were characterized using FT-IR, HRMS and NMR spectroscopy. Structure–activity relationships including reported compounds are discussed.
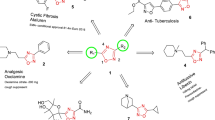
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neglected Tropical Diseases (NTDs) and infectious illnesses, such as malaria, tuberculosis and Zika fever, represent a major public health concern in many countries and regions worldwide, especially in developing ones [1]. Most of the drugs available for therapy are toxic and have considerable adverse effects. A considerable number is obsolete, especially with respect to resistance.
Trypanosoma brucei is one of the protozoan parasites that penetrates the blood–brain barrier causing injury associated with toxic effects of parasite-derived molecules or with immune response against infection. Other protozoan parasites that can cause pathology in the brain tropism include Toxoplasma, Plasmodium, Amoeba and, eventually, other Trypanosomatids such as T. cruzi and Leishmania. Together, these parasites affect billions of people worldwide and are responsible for more than 500,000 deaths annually [2]. New drugs against these parasitic protozoa are urgently needed to counteract drug resistance, toxicity and the high cost of commercially available drugs [3].
Recently, we reported about the synthesis of 1-benzyl [4, 5] and 1,3-dibenzyl derivatives of tetrahydropyridinylidene salts (THPS) [6]. Their activities against Trypanosoma brucei rhodesiense (T.b.r.), the protozoan pathogen of the East African form of sleeping sickness were investigated. Moreover, their activities against the sensitive NF54 strain and the multiresistant K1 strain of Plasmodium falciparum (P.falc.) were determined [6]. The most promising of these compounds were also investigated for their in vivo activity against Plasmodium berghei in a mouse model [4].
Due to the fact that compounds with electron withdrawing groups at the aromatic moiety show better and more selective activities, we introduced such groups in a series of new benzyl- and dibenzyl-THPS in order to reveal structure–activity relationships and to optimize the compounds regarding their activity and cytotoxicity.
Results and discussion
Chemistry
The synthesis of the benzyl-THPS 2–5 starts from bases 1a–1c [5, 7] by alkylation of the nitrogen atom of the dihydropyridine ring with benzyl halides as described earlier [4, 5]. The obtained compounds 2–5 have differing substitution in ring positions 4 of the piperidine as well as of the phenyl ring. Particularly THPS with electron-withdrawing substituents showed promising activity against protozoan parasites whereas electron-donating substituents like 4-methoxy or 4-alkyl showed moderate potency. Bigger alkyl groups in position 4 causes enhanced cytotoxicity and 3,4,5-trimethoxy compounds have low activity [5, 6]. Therefore, we prepared further analogues with cyano and nitro groups. In addition, the azepane moiety was chosen as a slightly more lipophilic amine component. Compounds 6–8 with an additional benzyl substituent in ring position 3 were afforded by reaction of benzyl-THPS 2–5 with benzyl halides in the presence of potassium carbonate. Analogues 9–12 with identical substitution in ring positions 1 and 3 were obtained by a one pot reaction of 1 with benzyl halides in the presence of potassium carbonate (Scheme 1).

The alkylation at the ring nitrogen in compounds 1 follows a SN2 mechanism. The electron-pair of the ring nitrogen attacks the carbon of the aryl–alkyl halide and leads to 13, the typical transition state. Detachment of the halide ion causes a migration of double bonds giving the N-benzyl-THPS 2–5 (Scheme 2).

The formation of the 1,3-dibenzyl compounds 6–8 starts with a proton abstraction in position 3 of compounds 2–5 to intermediate 14 using potassium carbonate as base. With the aryl–alkyl halide a SN2-type transition state 15 is supposed. By the leaving of the halide ion, 1,3-dibenzyl-THPS 6–8 are formed (Scheme 3).

Confirmation of structures
The formation of compounds 2–5 was verified by NMR spectroscopy. The successful alkylation was confirmed by the appearance of resonances of additional protons of the benzyl moiety between 4.77 and 4.95 ppm in 1H NMR spectra. For those protons long range couplings to C-2 and C-6 of the dihydropyridine ring were observed in HMBC spectra to establish connectivity. The additional proton signals of the aromatic protons of the benzyl residues are observed too in 1H NMR spectra. In 13C NMR spectra the signals for the methylene group of the benzyl residues appear at around 53 ppm. The additional signals for the aromatic carbons were detected as well. The results of the HRMS measurements confirmed the proposed structures too. Finally, the structures of compounds 2–5 are also established by a single X-ray crystal analysis of a reported compound prepared in the same manner [4].
The methylene protons of the additional benzyl group of compounds 6–12 appear as two doubledoublets at around 2.5 ppm (J = 13 and 10 Hz) and around 3 ppm (J = 13 and 5 Hz). The coupling with the smaller coupling constant is due to the interaction with the remaining proton in position 3, respectively. Furthermore, additional cross-peaks to H-3 in the H,H-COSY spectra, as well as long range couplings to C-4 in the HMBC spectra were observed to verify the position of the substitution. In 13C NMR spectra the signals for the methylene group attached to position 3 appear at around 35 ppm. The signal for C-3 shifted from around 39–45 ppm due to substitution and was detected in DEPT spectra as CH group instead of the former CH2 group. HRMS measurements confirmed the proposed structures of 6–12 as well. From a similar reported compound prepared by the same way, a single X-ray structure analysis is described [6].
Antiprotozoal activities
All new THPS were investigated for their activity against chloroquine sensitive P.falc. NF54 and T.b.r. as well as for their cytotoxicity against L-6 cells using microplate assays. The results are listed in Table 1. For comparative reasons known compounds were included. The most promising compounds with high antiplasmodial activity and high selectivity were additionally tested against P.falc. K1.
The majority of compounds 2–12 showed weak antitrypanosomal activity (IC50 = 4.42–156 µM) and low selectivity (SI = 0.20–37.8). Moderate activity against T.b.r. (IC50 = 0.51–1.65 µM) was observed for the new 4-nitrobenzyl compounds 8b and 8c. In comparison to their more active bis(4-chlorobenzyl) analogues 10a and 10b (IC50 L6 < 13 µM) they were less toxic (8b, 8c: IC50 L6 > 96 µM) and showed improved selectivity (SI = 122–190).
The new monobenzyl substituted compounds 2–5 exhibited moderate to good antiplasmodial activity (IC50 = 0.019–0.51 µM) against Plasmodium falciparum NF54. Their calculated selectivities were eminent (SI = 378–9207) except for compound 5c (SI = 77.9). Compounds 6–10 of the bisbenzyl series showed high activity (IC50 = 0.0014–0.043 µM) and outstanding selectivity (SI = 1525–99,524). Only the bis(4-nitrobenzyl) and the bis(4-cyanobenzyl) derivatives 11a and 12a were less active (IC50 = 0.18–0.97 µM) and less selective (SI = 210–1050).
A selection of promising compounds was also tested against the multiresistant K1 strain of P.falc.. The activities of compounds 3a, 7a, 7b, 9a and 9b (IC50 = 0.024–0.12 µM) were decreased in comparison to their activities against Plasmodium falciparum NF54 (IC50 = 0.0021–0.032 µM) as expected. Therefore, compound 7b, which was the most selective against the chloroquine sensitive NF54 strain (SI = 99,524), possessed still high but reduced selectivity against the K1 strain (SI = 6742). Interestingly, the activities of the 3-(4-nitrobenzyl) derivative 8b and its bis(4-chlorobenzyl) analogues 10a and 10b showed similar or increased activity against the multiresistant strain. Compounds 8b and 10a exhibited activity in the low nanomolar region (IC50 = 0.0016–0.006 µM), whereas 10b was even active in subnanomolar concentration (IC50 = 0.00038 µM). The selectivities of compounds 8b and 10b against this strain are excellent (SI = 29,211–33,500).
Structure–activity relationships
The above stated observation, that the presence of electron withdrawing substituents at the aromatic moiety increase the antiplasmodial activity was confirmed for compounds with only one benzyl residue attached to the ring nitrogen. Furthermore, due to the selection of 4-nitro and 4-cyano substituents, the selectivity was increased significantly due to the much lower cytotoxicity compared with 4-chloro substituted compounds. For the bis-benzyl substituted compounds we observed that the substitution pattern of 4-nitro and 4-cyano substituted benzyl residues in position 3 and at the same time unsubstituted benzyl residues at the ring nitrogen are advantageous for both, antiplasmodial activity and selectivity of compounds. As a result, compounds with outstanding activity and selectivity were yielded. The introduction of 4-nitro and 4-cyano substituted benzyl residues to both positions resulted in lower potent compounds.
The insertion of the bigger azepane ring as amino moiety in position 4 was in general advantageous, often the compounds with this substitution were the most active within their series.
Conclusion
To continue our studies about the antiprotozoal activities of tetrahydropyridinylidene ammonium salts, we prepared 1-substituted benzyl and 1,3-disubstituted dibenzyl derivatives with electron withdrawing substituents at the aromatic moieties, since such substituents were identified to be advantageous for antiplasmodial action. 4-Cyano and 4-nitro compounds show increased antiplasmodial potency and raised selectivity due to less cytotoxicity compared to 4-chloro compounds. In addition to that, the larger azepane ring was introduced as an amino substituent in ring position 4 giving highly active compounds. The most promising of the new compounds is the 1-benzyl-3-(4-nitrobenzyl) derivative 8b showing low cytotoxicity and antiplasmodial activity against a sensitive and a multiresistant strain of Plasmodium falciparum in low nanomolar concentration. The main goal, to improve the biological activity and to decrease cytotoxicity was reached. Physicochemical properties where calculated and a correlation between biological activities and lipophilicity of compounds was detected. Further investigations should provide insight into structure–activity relationships of these compounds and varying substitution pattern on the aromatic moieties.
Experimental
Melting points were obtained on a digital melting point apparatus Electrothermal IA 9200. IR spectra: Bruker Alpha Platinum ATR FT-IR spectrometer (KBr discs). NMR spectra: Bruker Ascend 400, 5 mm tubes, spectra were acquired in CDCl3 containing 0.03% TMS. Chemical shifts were recorded in parts per million (ppm), for 1H spectra TMS (0.00 ppm) was used as internal standard and for 13C spectra the central peak of the CDCl3 signal was used as the internal reference (77.0 ppm). Some spectra were acquired in DMSO-d6. In this case the central peaks of the DMSO-d5 signal at 2.49 ppm in 1H spectra and at 39.7 ppm in 13C spectra served as internal reference. Abbreviations: aromatic H, ArH; aromatic C, ArC, quaternary aromatic C, ArCq. Signal multiplicities are abbreviated as follows: s, singlet; d, doublet; dd, doubledoublet; ddd, doubledoubledoublet; dt, doubletriplet; t, triplet; m, multiplet; br, broad. Coupling constants (J) are reported in Hertz (Hz). 1H and 13C resonances were assigned using 1H,1H- and 1H,13C-correlation spectra. 1H and 13C resonances are numbered as given in the formulae. HR-MS: Micromass tofspec 3E spectrometer (MALDI), GCT-Premier, Waters (EI, 70 eV), Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer, Thermo Fisher Scientific (HESI, 3.5 kV). Materials: column chromatography (CC): silica gel 60 (Merck 70–230 mesh, pore-diameter 0.6 nm), aluminium oxide (Alox) basic (Fluka for chromatography, 0.05–0.15 mm, Brockmann activity I, basic); Alox neutral 90 (Merck, 0.063–0.2 mm, activity I, neutral); thin-layer chromatography (TLC): TLC plates (Merck, silica gel 60 F254 0.2 mm, 200 × 200 mm); TLC plates (Merck, Alox 60 F254 neutral, 200 × 200 mm); the substances were detected in UV light at 254 nm. If no stationary phase is mentioned (CC and TLC) the separation took place using silica gel. The preparation of benzyl compounds 2a, 2b, 4a, 4b, 5a, 5b is reported [4, 5] as well as the synthesis of dibenzyl compounds 6a, 7a, 8a, 9a, 9b, 10a and 10b [6].
N-(1-Benzyl-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene)azepan-1-ium bromide (2c, C20H29BrN2)
A solution of 1.159 g of 1c (5.62 mmol) and 1.604 g of benzyl bromide (9.38 mmol) in 25 cm3 of CHCl3 was stirred at r.t. for 20 h. A part of the solvent was evaporated in vacuo and cooled with an ice bath. Ethyl acetate was added until crystallisation seemed to be complete. The solid was sucked off and dried giving 1.540 g (73%) of 1c as bright yellow crystals. Rf = 0.31 (CH2Cl2:MeOH = 9:1); m.p.: 180 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 1.22 (s, 6H, 2CH3), 1.51 (br, s, 4H, H-3′), 1.66–1.76 (m, 4H, H-2′), 2.97 (s, 2H, H-3), 3.73 (td, J = 5.2, 1.8 Hz, 4H, H-1′), 4.77 (s, 2H, ArCH2), 5.43 (d, J = 7.0 Hz, 1H, H-5), 7.30–7.43 (m, 5H, ArH), 7.80 (d, J = 7.0 Hz, 1H, H-6) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.24 (2CH3), 25.34, 25.45, 25.68 (C-2′, C-3′), 27.98 (C-2′), 38.55 (C-3), 51.34, 51.66 (C-1’), 53.58 (ArCH2), 57.57 (C-2), 87.58 (C-5), 127.58, 127.99, 128.94 (ArC), 137.40 (ArCq), 157.51 (C-6), 164.74 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2924, 1558, 1505, 1446, 1397, 1373, 1351, 1231, 1106, 722 cm−1; HRMS (EI+): m/z calcd. for C20H28N2 ([M-HBr]+) 296.2253, found 296.2255.
N-[1-(4-Cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]pyrrolidin-1-ium bromide (3a, C19H24BrN3)
A solution of 732 mg of 1a (4.11 mmol) and 1.382 g of 4-bromomethyl benzonitrile (6.98 mmol) in 20 cm3 of CHCl3 was stirred at r.t. for 4 d. Then ethyl acetate was added with cooling on an ice bath until precipitation seemed complete. The solid was sucked off and dried giving 1.521 g (99%) of 3a as yellowish crystals. Rf = 0.20 (CH2Cl2:MeOH = 8:1); m.p.: 172 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 1.21 (s, 6H, 2CH3), 1.93–2.00 (m, 4H, 2CH2), 2.95 (s, 2H, H-3), 3.55 (t, J = 6.2 Hz, 2H, NCH2), 3.67 (t, J = 6.2 Hz, 2H, NCH2), 4.85 (s, 2H, ArCH2), 5.30 (d, J = 7.0 Hz, 1H, H-5), 7.54 (d, J = 8.1 Hz, 2H, ArH), 7.78 (d, J = 7.0 Hz, 1H, H-6), 7.88 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.50 (2CH3), 24.31, 24.54 (2CH2), 40.30 (C-3), 49.80, 50.00 (2NCH2), 53.10 (ArCH2), 57.46 (C-2), 88.98 (C-5), 110.54 (ArCq), 118.76 (CN), 128.13, 132.78 (ArC), 143.89 (ArCq), 157.66 (C-6), 162.60 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2931, 2225, 1607, 1550, 1479, 1441, 1407, 1336, 1272, 1235, 1187, 1106, 999, 969, 860, 831, 766 cm−1; HRMS (EI+): m/z calcd. for C19H23N3 ([M-HBr]+) 293.1892, found 293.1886.
N-[1-(4-Cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]piperidin-1-iumbromide (3b, C20H26BrN3)
A solution of 1.171 g of 1b (6.09 mmol) and of 2.05 g 4-bromomethyl benzonitrile (10.4 mmol) in 30 cm3 of CHCl3 was stirred at r.t. for 5 d. The solvent was evaporated in vacuo and the residue dissolved in CHCl3. The hot solution was treated with charcoal, filtered *and ethyl acetate was added dropwise until first turbidity appeared. Then it was stirred at r.t. until the precipitation seemed to be complete. The precipitate was sucked off and dried giving 1.75 g of 3b (74%) as yellowish crystals. Rf = 0.22 (CH2Cl2:MeOH = 8:1); m.p.: 133 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 1.18 (s, 6H, 2CH3), 1.58–1.73 (m, 6H, 3CH2), 2.97 (s, 2H, H-3), 3.67–3.72 (m, 4H, 2NCH2), 4.85 (s, 2H, ArCH2), 5.58 (d, J = 7.0 Hz, 1H, H-5), 7.55 (d, J = 7.3 Hz, 2H, ArH), 7.78 (d, J = 6.2 Hz, 1H, H-6), 7.87 (d, J = 6.6 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.32 (CH2, 2CH3), 25.95, 26.91 (2CH2), 38.62 (C-3), 49.34, 49.40 (2NCH2), 52.99 (ArCH2), 57.56 (C-2), 88.03 (C-5), 110.52 (ArCq), 118.79 (CN), 128.22, 132.75 (ArC), 143.77 (ArCq), 157.97 (C-6), 163.65 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2941, 2860, 2225, 1608, 1558, 1468, 1444, 1403, 1353, 1254, 1238, 1186, 1111, 1018, 951, 859, 736 cm−1; HRMS (EI+): m/z calcd. for C20H25N3 ([M-HBr]+) 307.2048, found 307.2054.
N-[1-(4-Cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]azepan-1-ium bromide (3c, C21H28BrN3)
A solution of 1.763 g of 1c (8.54 mmol) and 2.798 g of 4-bromomethyl benzonitrile (14.27 mmol) in 45 cm3 of CHCl3 was stirred at r.t. for 7 d. A part of the solvent was evaporated in vacuo and ethyl acetate was added with stirring and cooling until crystallization seemed to be complete. The precipitate was sucked off and dried giving 3.092 g of 3c (90%) as bright yellow crystals. Rf = 0.32 (CH2Cl2: MeOH = 10:1); m.p.: 135 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 1.20 (s, 6H, 2CH3), 1.53 (br, s, 4H, H-3′), 1.64–1.79 (m, 4H, H-2′), 3.01 (s, 2H, H-3), 3.75 (t, J = 6.1 Hz, 4H, H-1 ‘), 4.89 (s, 2H, ArCH2), 5.48 (d, J = 7.1 Hz, 1H, H-5), 7.57 (d, J = 8.1 Hz, 2H, ArH), 7.82 (d, J = 7.1 Hz, 1H, H-6), 7.88 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.17 (2CH3), 25.38, 25.41, 25.64 (C-2′, C-3′), 27.88 (C-2′), 38.47 (C-3), 51.46, 51.79 (C-1′), 53.11 (ArCH2), 57.70 (C-2), 88.04 (C-5), 110.61 (ArCq), 118.80 (CN), 128.26, 132.80 (ArC), 143.66 (ArCq), 157.99 (C-6), 165.11 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2929, 2227, 1608, 1557, 1401, 1350, 1234, 1184, 1107 cm−1; HRMS (HESI): m/z calcd. for C21H28N3 ([M-Br]+) 322.2282, found 322.2274.
N-[2,2-Dimethyl-1-(4-nitrobenzyl)-1,2,3,4-tetrahydropyridin-4-ylidene]azepan-1-ium bromide (4c, C20H28BrN3O2)
A solution of 1.073 g of 1c (5.2 mmol) and 1.876 g of 4-nitrobenzyl bromide (8.68 mmol) in 25 cm3 of CHCl3 was stirred at r.t. for 20 h. A part of the solvent was evaporated in vacuo and ethyl acetate was added with stirring and cooling until crystallization was completed. The solid was sucked off giving 1.692 g of 4c (77%) as a yellow powder which still contained CHCl3 after rigorous drying. Therefore, it was dissolved in CH2Cl2 and the solvent was evaporated in vacuo. After drying in vacuo a yellow foam without any traces of CHCl3 was yielded. Rf = 0.35 (CH2Cl2: MeOH = 9:1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.21 (s, 6H, 2CH3), 1.53 (br, s, 4H, H-3′), 1.62–1.82 (m, 4H, H-2′), 3.02 (s, 2H, H-3), 3.76 (t, J = 6.1 Hz, 4H, H-1′), 4.95 (s, 2H, ArCH2), 5.50 (d, J = 7.1 Hz, 1H, H-5), 7.65 (d, J = 8.3 Hz, 2H, ArH), 7.86 (d, J = 7.1 Hz, 1H, H-6), 8.25 (d, J = 8.3 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.16 (2CH3), 25.39, 25.41, 25.64 (C-2′, C-3′), 27.87 (C-2′), 38.49 (C-3), 51.49, 51.82 (C-1′), 52.93 (ArCH2), 57.75 (C-2), 88.13 (C-5), 123.96, 128.54 (ArC), 145.81, 147.13 (ArCq), 158.04 (C-6), 165.16 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2930, 1609, 1559, 1519, 1398, 1343, 1106 cm−1; HRMS (HESI): m/z calcd. for C20H28N3O2 ([M-Br]+) 342.2182, found 342.2171.
N-[1-(4-Chlorobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]azepan-1-ium chloride (5c, C20H28Cl2N2)
A solution of 200 mg of 1c (0.96 mmol) and 310 mg of 4-chlorobenzyl bromide (1.93 mmol) in 1.5 cm3 of CHCl3 was stirred at r.t. for 3 d. The solvent was evaporated and the residue dissolved in hot ethanol, treated with charcoal and filtered. The solvent was removed in vacuo and the pure product precipitated from a mixture of CHCl3 and ethyl acetate. It was sucked off and dried giving 320 mg of 5c (91%) as grey precipitate. For analytical purposes it was dissolved in CHCl3 and ethyl acetate was added. The product crystallized overnight in form of grey platelets. Rf = 0.13 (CH2Cl2:MeOH = 8:1); m.p.: 132 °C; 1H NMR (CDCl3, 400 MHz): δ = 1.21 (s, 6H, 2CH3), 1.51 (br, s, 4H, H-3′), 1.67–1.74 (m, 4H, H-2′), 2.98 (s, 2H, H-3), 3.71–3.75 (m, 4H, H-1′), 4.79 (s, 2H, ArCH2), 5.43 (d, J = 7.0 Hz, 1H, H-5), 7.40 (d, J = 8.4 Hz, 2H, ArH), 7.45 (d, J = 8.4 Hz, 2H, ArH), 7.88 (d, J = 7.0 Hz, 1H, H-6) ppm; 13C NMR (CDCl3, 100 MHz): δ = 23.21 (2CH3), 25.34, 25.42, 25.65 (C-2′, C-3′), 27.92 (C-2′), 38.54 (C-3), 51.35, 51.67 (C-1′), 52.81 (ArCH2), 57.61 (C-2), 87.72 (C-5), 128.80, 129.51 (ArC), 132.49, 136.63 (ArCq), 157.70 (C-6), 164.82 (C-4) ppm; IR (KBr): \(\overline{V}\) = 3426, 2926, 1557, 1445, 1401, 1237, 1178, 1106, 759 cm−1; HRMS (EI+): m/z calcd. for C20H27ClN2 ([M-HCl]+) 330.1863, found 330.1852.
(3RS)-(±)-N-[1-Benzyl-3-(4-cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]piperidin-1-ium bromide (7b, C27H32BrN3)
A mixture of 1.007 g 2b (2.77 mmol) and 654 mg of 4-bromomethyl benzonitrile (3.33 mmol) in 50 cm3 of CHCl3 was refluxed overnight in the presence of 3.2 g K2CO3 (23.2 mmol). Charcoal was added to the mixture and then it was heated until it boiled. It was filtered and the solvent was evaporated in vacuo. The residue was dissolved in acetone and ethyl acetate was added until the mixture got turbid. Upon stirring at r.t. a precipitate was formed which was sucked off and was recrystallized from acetone, giving 7b as beige crystals containing acetone. Therefore, it was dissolved in CH2Cl2 and the solvent was evaporated in vacuo. After drying in vacuo 243 mg (18%) of a solvent-free beige foam was yielded. Rf = 0.36 (CH2Cl2:MeOH = 8:1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.06 (br, s, 4H, CH3, CH2), 1.16–1.32 (m, 1H, CH2), 1.34–1.49 (m, 5H, CH3, CH2), 1.59–1.62 (m, 2H, CH2), 2.36 (br, dt, J = 13.0, 2.7 Hz, 1H, NCH2), 2.49–2.54 (m, 1H, ArCH2CH), 3.04 (dd, J = 13.2, 5.1 Hz, 1H, ArCH2CH), 3.09 (br, t, J = 10.3 Hz, 1H, NCH2), 3.46–3.56 (m, 2H, H-3, NCH2), 3.84 (br, d, J = 12.8 Hz, 1H, NCH2), 4.72 (d, J = 15.4 Hz, 1H, ArCH2N), 4.84 (d, J = 15.4 Hz, 1H, ArCH2N), 5.54 (d, J = 6.6 Hz, 1H, H-5), 7.33–7.44 (m, 7H, ArH), 7.74 (d, J = 8.1 Hz, 2H, ArH), 7.87 (d, J = 7.0 Hz, 1H, H-6) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.30, 22.13 (2CH3), 22.83, 25.94, 27.11 (3CH2), 34.71 (ArCH2CH), 44.94 (C-3), 49.18, 49.51 (N(CH2)2), 53.86 (ArCH2N), 60.77 (C-2), 86.60 (C-5), 109.89 (ArCq), 118.97 (CN), 128.11, 128.15, 128.93, 130.82, 132.19 (ArC), 137.05, 143.35 (ArCq), 156.14 (C-6), 167.12 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2950, 2222, 1604, 1557, 1479, 1453, 1397, 1349, 1298, 1231, 1162, 1097, 1017, 947, 829, 754, 719 cm−1; HRMS (EI+): m/z calcd. for C27H31N3 ([M-HBr]+) 397.2518, found 397.2535.
(3RS)-(±)-N-[1-Benzyl-3-(4-cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]azepan-1-ium bromide (7c, C28H34BrN3)
A mixture of 1 g of 2c (2.65 mmol) and 623 mg of 4-bromomethyl benzonitrile (3.18 mmol) in 55 cm3 of CHCl3 was refluxed overnight in the presence of 3.392 g of K2CO3 (24.54 mmol). It was cooled down to r.t., then 55 cm3 of CHCl3 were added and it was treated with charcoal and filtered. The solvent was evaporated in vacuo giving a brown resin. It was dissolved in acetone. Upon addition of a few drops of ethyl acetate crystallization occurred giving brown crystals which were sucked off and dried. The crystals contained acetone. The solid was dissolved in CH2Cl2 and evaporated in vacuo giving 172 mg 7c (13%) as brown foam. Rf = 0.89 (CH2Cl2:MeOH = 10:1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.10 (s, 3H, CH3), 1.17–1.62 (m, 7H, H-2′, H-3′), 1.41 (s, 3H, CH3), 1.66–1.81 (m, 1H, H-2′), 2.22–2.46 (m, 1H, H-1′), 2.55 (dd, J = 13.1, 10.3 Hz, 1H, ArCH2CH), 3.06 (dd, J = 12.9, 5.3 Hz, 1H, ArCH2CH), 3.16 (ddd, J = 13.8, 10.2, 3.9 Hz, 1H, H-1′), 3.24–3.38 (m, 2H, H-1′, H-3), 3.78 (dt, J = 14.1, 4.3 Hz, 1H, H-1′), 4.72 (d, J = 15.5 Hz, 1H, ArCH2N), 4.83 (d, J = 15.5 Hz, 1H, ArCH2N), 5.44 (d, J = 6.9 Hz, 1H, H-5), 7.33–7.57 (m, 7H, ArH), 7.75 (d, J = 8.1 Hz, 2H, ArH), 7.83 (d, J = 6.9 Hz, 1H, H-6) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.55, 22.18 (2CH3), 24.73, 26.02, 29.13 (C-2′, C-3′), 34.82 (ArCH2CH), 45.49 (C-3), 50.97, 51.25 (C-1′), 54.02 (ArCH2N), 60.97 (C-2), 87.33 (C-5), 110.01 (ArCq), 119.06 (CN), 128.18, 128.27, 129.04, 130.85, 132.15 (ArC), 136.99, 143.37 (ArCq), 156.40 (C-6), 167.92 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2934, 2223, 1557, 1453, 1401, 1352, 1231, 1094, 763 cm−1; HRMS (EI+): m/z calcd. for C28H33N3 ([M-HBr]+) 411.2675, found 411.2689.
(3RS)-(±)-N-[1-Benzyl-2,2-dimethyl-3-(4-nitrobenzyl)-1,2,3,4-tetrahydropyridin-4-ylidene]piperidin-1-ium bromide (8b, C26H32BrN3O2)
A mixture of 1048 mg of 2b (2.88 mmol) and 749 mg of 4-nitrobenzyl bromide (3.47 mmol) in 50 cm3 of CHCl3 was refluxed in the presence of 3.24 g of K2CO3 (23.54 mmol) overnight. It was treated with charcoal filtered and the solvent evaporated in vacuo. The residue was dissolved with acetone and ethyl acetate was added until the mixture got turbid. Upon stirring at r.t. a precipitate was formed which was sucked off and recrystallized from acetone, giving 8b as beige crystals containing acetone. Therefore, the crystals were dissolved in CH2Cl2 and the solvent was evaporated to yield 282 mg of 8b (20%) as beige foam. Rf = 0.24 (CH2Cl2:MeOH = 8:1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.07 (s, 3H, CH3), 1.11–1.24 (m, 2H, CH2), 1.36–1.42 (m, 2H, CH2), 1.46 (s, 3H, CH3), 1.50–1.61 (m, 2H, CH2), 2.34 (ddd, J = 13.2, 10.2, 2.9 Hz, 1H, NCH2), 2.57 (dd, J = 12.6, 11.0 Hz, 1H, ArCH2CH), 3.05 (br, t, J = 10.3 Hz, 1H, NCH2), 3.10 (dd, J = 12.6, 5.1 Hz, 1H, ArCH2CH), 3.53–3.57 (m, 2H, H-3, NCH2), 3.86–3.89 (m, 1H, NCH2), 4.73 (d, J = 15.6 Hz, 1H, ArCH2N), 4.85 (d, J = 15.6 Hz, 1H, ArCH2N), 5.55 (d, J = 7.0 Hz, 1H, H-5), 7.32–7.45 (m, 7H, ArH), 7.88 (d, J = 7.0 Hz, 1H, H-6), 8.13 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.30, 22.12 (2CH3), 22.79, 26.00, 27.31 (3CH2), 34.41 (ArCH2CH), 44.93 (C-3), 49.16, 49.56 (2NCH2), 53.87 (ArCH2N), 60.76 (C-2), 86.62 (C-5), 123.33, 128.12, 128.15, 128.93, 131.03 (ArC), 137.03, 145.57, 146.67 (ArCq), 156.16 (C-6), 166.94 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2940, 1603, 1556, 1517, 1452, 1396, 1345, 1234, 1164, 1108, 1016, 859, 703 cm−1; HRMS (EI+): m/z calcd. for C26H31N3O2 ([M-HBr]+) 417.2416, found 417.2428.
(3RS)-(±)-N-[1-Benzyl-2,2-dimethyl-3-(4-nitrobenzyl)-1,2,3,4-tetrahydropyridin-4-ylidene]azepan-1-ium bromide (8c, C27H34BrN3O2)
A mixture of 1.2 g of 2c (3.2 mmol) and 824 mg of 4-nitrobenzyl bromide (3.8 mmol) in 66 cm3 CHCl3 was stirred for 60 d at r.t. in the presence of 4.095 g of K2CO3 (29.6 mmol). 66 cm3 of CHCl3 were added and it was treated with charcoal and filtered. The solvent was evaporated in vacuo and the residue was dissolved in MeOH, diluted with water and was put into a separatory funnel. The aqueous layer was extracted three times with ether. The combined ethereal layers were discarded and the aqueous layer was extracted five times with CH2Cl2. The organic layer was treated with anhydrous Na2SO4 and filtered. The solvents were evaporated in vacuo and the residue crystallized from ethyl acetate giving a brown powder which was dissolved in CH2Cl2 and evaporated in vacuo giving 113 mg of 8c (0.22 mmol, 7%) as a brown foam. Rf = 0.34 (CH2Cl2: MeOH = 10:1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.10 (s, 3H, CH3), 1.20–1.79 (m, 8H, H-2′, H-3′), 1.43 (s, 3H, CH3), 2.29–2.40 (m, 1H, H-1′), 2.56–2.67 (m, 1H, ArCH2CH), 3.08–3.19 (m, 2H, H-1′, ArCH2CH), 3.27–3.40 (m, 2H, H-1′, H-3), 3.69 − 3.82 (m, 1H, H-1′), 4.73 (d, J = 15.5 Hz, 1H, ArCH2N), 4.83 (d, J = 15.4 Hz, 1H, ArCH2N), 5.44 (d, J = 7.1 Hz, 1H, H-5), 7.31–7.47 (m, 7H, ArH), 7.85 (d, J = 6.9 Hz, 1H, H-6), 8.13 (d, J = 8.3 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.54, 22.14 (2CH3), 24.68, 26.00, 29.18 (C-2′, C-3′), 34.50 (ArCH2CH), 45.50 (C-3), 50.92, 51.25 (C-1′), 54.01 (ArCH2N), 60.95 (C-2), 87.35 (C-5), 123.28, 128.18, 128.25, 129.01, 131.07 (ArC), 136.91, 145.57, 146.73 (ArCq), 156.38 (C-6), 167.75 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2927, 1717, 1556, 1519, 1455, 1396, 1345, 1108 cm−1; HRMS (HESI): m/z calcd. for C27H34N3O2 ([M-Br]+) 432.2651, found 432.2639.
(3RS)-(±)-N-(1,3-Dibenzyl-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene)azepan-1-ium bromide (9c, C27H35BrN2)
A mixture of 1 g of 1c (2.65 mmol) and of 544 mg benzyl bromide (3.18 mmol) in 55 cm3 of CHCl3 was refluxed overnight in the presence of 3.392 g of K2CO3 (24.54 mmol). It was cooled down to r.t. and 55 cm3 of CHCl3 were added and then treated with charcoal and filtered. The solvent was evaporated in vacuo giving a residue which was dissolved in acetone. Crystallization gave a brown powder which was sucked off. The solid was dissolved in CH2Cl2 and evaporated in vacuo giving 417 mg of 9c (56%) as brownish foam. Rf = 0.93 (CH2Cl2: MeOH = 9:1); 1H NMR (CDCl3, 400 MHz): δ = 1.24 (s, 3H, CH3), 1.30–1.62 (m, 7H, H-2′, H-3′), 1.67 (s, 3H, CH3), 1.80–1.91 (m, 1H, H-2′), 2.16 (ddd, J = 14.0, 8.9, 4.5 Hz, 1H, H-1′), 2.56 (dd, J = 12.8, 10.9 Hz, 1H, ArCH2CH), 3.06 (ddd, J = 13.6, 9.3, 3.6 Hz, 1H, H-1′), 3.15 (dd, J = 12.8, 5.1 Hz, 1H, ArCH2CH), 3.50 (dd, J = 10.9, 5.1 Hz, 1H, H-3), 3.64 (dt, J = 14.4, 5.1 Hz, 1H, H-1′), 3.73 (dt, J = 14.4, 5.0 Hz, 1H, H-1′), 4.72 (d, J = 15.2 Hz, 1H, ArCH2N), 4.82 (d, J = 15.2 Hz, 1H, ArCH2N), 5.45 (d, J = 6.9 Hz, 1H, H-5), 7.23–7.39 (m, 10H, ArH), 8.05 (d, J = 6.9 Hz, 1H, H-6) ppm; 13C NMR (CDCl3, 100 MHz): δ = 21.98, 22.80 (2CH3), 25.10, 25.16, 26.48, 29.18 (C-2′, C-3′), 35.12 (ArCH2CH), 47.11 (C-3), 51.22, 51.71 (C-1′), 54.79 (ArCH2N), 61.22 (C-2), 87.52 (C-5), 127.09, 128.06, 128.28, 128.40, 128.98, 129.66 (ArC), 135.27, 136.43 (ArCq), 155.87 (C-6), 168.65 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2926, 1555, 1403, 1351, 1242, 1153, 1096, 763 cm−1; HRMS (EI+): m/z calcd. for C27H34N2 ([M-HBr]+) 386.2722, found 386.2740.
(3RS)-(±)-N-[2,2-Dimethyl-1,3-bis(4-nitrobenzyl)-1,2,3,4-tetrahydropyridin-4-ylidene]pyrrolidin-1-ium bromide (11a, C25H29BrN4O4)
To a solution of 1 g of 1a (5.64 mmol) in 25 cm3 of CHCl3 2.03 g of 4-nitrobenzyl bromide (9.4 mmol) were added. It was stirred at r.t. for two days and ethyl acetate was added to the already turbid solution. The formed precipitate was sucked off giving 1.98 g (89%) of the monosubstituted product 4a. 950 mg of 4a (2.41 mmol) and 627 mg of 4-nitrobenzyl bromide (2.9 mmol) in 40 cm3 of CHCl3 were refluxed overnight in the presence of 2.7 g of K2CO3 (19.5 mmol). Then 100 cm3 of CHCl3 were added and the mixture was treated with charcoal and filtered. The solvent was evaporated in vacuo and the residue was dissolved in acetone and ethyl acetate was added until the solution got turbid. Upon stirring on an ice bath, a precipitate was formed which was sucked off and washed with ethyl acetate. It was recrystallized three times from acetone giving bright brown needles containing acetone. They were sucked off and dissolved in CH2Cl2. The solvent was evaporated to yield 120 mg of 11a (4%) as a brownish foam. Rf = 0.13 (CH2Cl2:MeOH = 8:1); 1H NMR (DMSO-d6, 400 MHz): δ = 10.17 (s, 3H, CH3), 1.22–1.30 (m, 1H, CH2), 1.34 (s, 3H, CH3), 1.56–1.69 (m, 2H, CH2), 1.82 (quin, J = 6.1 Hz, 1H, CH2), 2.26 (quin, J = 6.5 Hz, 1H, NCH2), 2.69 (dd, J = 12.1, 10.6 Hz, 1H, ArCH2CH), 3.20 (dd, J = 12.8, 5.1 Hz, 1H, ArCH2CH), 3.33–3.43 (m, 2H, H-3, NCH2), 3.50–3.62 (m, 2H, NCH2), 4.90 (d, J = 16.1 Hz, 1H, ArCH2N), 5.05 (d, J = 16.5 Hz, 1H, ArCH2N), 5.34 (d, J = 7.0 Hz, 1H, H-5), 7.48 (d, J = 8.4 Hz, 2H, ArH), 7.76 (d, J = 8.1 Hz, 2H, ArH), 8.00 (d, J = 7.0 Hz, 1H, H-6), 8.14 (d, J = 8.4 Hz, 2H, ArH), 8.27 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.12, 22.35 (2CH3), 23.81, 24.36 (2CH2), 34.28 (ArCH2CH), 47.49 (C-3), 50.01, 50.17 (2NCH2), 53.20 (ArCH2N), 61.32 (C-2), 88.43 (C-5), 123.40, 124.05, 129.19, 131.06 (ArC), 145.31, 145.95, 146.66, 147.26 (ArCq), 156.62 (C-6), 166.06 (C-4) ppm; IR (KBr): \(\overline{V}\) = 2940, 1606, 1560, 1518, 1449, 1394, 1346, 1230, 1164, 1108, 859, 752 cm−1; HRMS (EI+): m/z calcd. for C24H25N4O4 ([M-HBr-CH3]+) 433.1876, found 433.1887.
(3RS)-(±)-N-[1,3-Bis(4-cyanobenzyl)-2,2-dimethyl-1,2,3,4-tetrahydropyridin-4-ylidene]pyrrolidin-1-ium bromide (12a, C27H29BrN4)
A mixture of 1.615 g of 1a (9.06 mmol) and 3.919 g of 4-bromomethyl benzonitrile (19.99 mmol) in 50 cm3 of CHCl3 was stirred for 6 d at r.t. in the presence of 4.673 g of K2CO3 (33.81 mmol). The reaction mixture was treated with charcoal, filtered and the solvent was evaporated in vacuo. The residue was dissolved in acetone and ethyl acetate was added until the first turbidity appeared. While stirring at r.t. and then on an ice-bath a precipitate was formed which was sucked of and recrystallized from acetone giving a mixture of bis- and monosubstituted products as white powder. In order to complete the reaction, the mixture was dissolved in 35 cm3 of CHCl3 and refluxed overnight in the presence of 2.215 g of K2CO3 (16.03 mmol) and 996 mg 4-bromomethyl benzonitrile (5.08 mmol). Then 15 cm3 of CHCl3 were added and the mixture was treated with charcoal and filtered. The solvent was evaporated in vacuo and the residue was dissolved in acetone. Then ethyl acetate was added until the first turbidity appeared. While stirring at r.t. and then on an ice-bath a precipitate was formed which was sucked of, recrystallized from acetone and dried giving 0.817 g of 12a (18%) as white powder containing acetone. Therefore, it was dissolved in CHCl3 and the solvent was evaporated to yield a white foam. Rf = 0.34 (CH2Cl2:MeOH = 9: 1); 1H NMR (DMSO-d6, 400 MHz): δ = 1.12 (s, 3H, CH3), 1.25 (quin, J = 6.3 Hz, 1H, CH2), 1.29 (s, 3H, CH3), 1.57–1.73 (m, 2H, CH2), 1.78–1.86 (m, 1H, CH2), 2.24 (quin, J = 6.5 Hz, 1H, NCH2), 2.62 (dd, J = 12.9, 9.9 Hz, 1H, ArCH2CH), 3.07 (dd, J = 12.9, 5.4 Hz, 1H, ArCH2CH), 3.16 (dd, J = 10.1, 5.5 Hz, 1H, H-3), 3.28–3.36 (m, 1H, NCH2), 3.47–3.55 (m, 2H, NCH2), 4.79 (d, J = 16.1 Hz, 1H, ArCH2N), 4.91 (d, J = 16.1 Hz, 1H, ArCH2N), 5.30 (d, J = 6.8 Hz, 1H, H-5), 7.37 (d, J = 8.0 Hz, 2H, ArH), 7.64 (d, J = 8.0 Hz, 2H, ArH), 7.77 (d, J = 8.0 Hz, 2H, ArH), 7.87 (d, J = 6.8 Hz, 1H, H-6), 7.91 (d, J = 8.0 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.14, 22.36 (2CH3), 23.84, 24.37 (2CH2), 34.51 (ArCH2CH), 47.54 (C-3), 49.96, 50.06 (2NCH2), 53.49 (ArCH2N), 61.24 (C-2), 88.34 (C-5), 109.92, 110.86 (ArCq), 118.78, 118.89 (CN), 128.89, 130.78, 132.29, 132.91 (ArC), 143.21, 143.72 (ArCq), 156.58 (C-6), 166.06 (C-4) ppm; IR (KBr): \(\overline{V}\)= 2925, 2227, 1608, 1558, 1448, 1395, 1333, 1230, 1163 cm−1; HRMS (HESI): m/z calcd. for C27H29N4 ([M-Br]+) 409.2392, found 409.2380.
In vitro growth inhibition assay of Plasmodium falciparum NF54
In vitro activity against erythrocytic stages of P. falciparum was determined using a 3H-hypoxanthine incorporation assay [8, 9], using the drug sensitive NF54 strain (Schipol Airport, The Netherlands, [10]) and the standard drug chloroquine (Sigma C6628). Compounds were dissolved in DMSO at 10 mg/cm3 and added to parasite cultures incubated in RPMI 1640 medium without hypoxanthine, supplemented with HEPES (5.94 g/dm3), NaHCO3 (2.1 g/dm3), neomycin (100 U/cm3), AlbumaxR (5 g/dm3) and washed human red cells A+ at 2.5% haematocrit (0.3% parasitaemia). Serial drug dilutions of eleven threefold dilution steps covering a range from 100 to 0.002 μg/cm3 were prepared. The 96-well plates were incubated in a humidified atmosphere at 37 °C; 4% CO2, 3% O2, 93% N2. After 48 h 50 mm3 of 3H-hypoxanthine (= 0.5 μCi) was added to each well of the plate. The plates were incubated for a further 24 h under the same conditions. The plates were then harvested with a Betaplate™ cell harvester (Wallac, Zurich, Switzerland), and the red blood cells were transferred onto a glass fibre filter and then washed with distilled water. The dried filters were inserted into a plastic foil with 10 cm3 of scintillation fluid and counted in a Betaplate™ liquid scintillation counter (Wallac, Zurich, Switzerland). IC50 values were calculated from sigmoidal inhibition curves by linear regression [11] using Microsoft Excel. Chloroquine was used as control.
In vitro growth inhibition assay of Trypanosoma b. rhodesiense
Minimum Essential Medium (50 mm3) supplemented according to [12] with 25 mM HEPES, 1 g/dm3 additional glucose, 1% MEM non-essential amino acids (100x), 0.2 mM 2-mercaptoethanol, 1 mM Na-pyruvate and 15% heat inactivated horse serum was added to each well of a 96-well microtiter plate. Serial drug dilutions of eleven threefold dilution steps covering a range from 100 to 0.002 μg/cm3 were prepared. Then 4 × 103 bloodstream forms of T. b. rhodesiense (STIB 900) in 50 mm3 was added to each well and the plate incubated at 37 °C under a 5% CO2 atmosphere for 70 h. 10 mm3 Alamar Blue (resazurin, 12.5 mg in 100 cm3 double-distilled water) was then added to each well and incubation continued for a further 2–4 h [13]. Then the plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. The IC50 values were calculated by linear regression [11] from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Cooperation, Sunnyvale, CA, USA). Melarsoprol was used as control.
Cytotoxicity against L6-cells
Assays were performed in 96-well microtiter plates, each well containing 100 mm3 of RPMI 1640 medium supplemented with 1% L-glutamine (200 mM) and 10% fetal bovine serum, and 4000 L-6 cells (a primary cell line derived from rat skeletal myoblasts) [14, 15]. Serial drug dilutions of eleven threefold dilution steps covering a range from 100 to 0.002 μg/cm3 were prepared. After 70 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterile conditions. 10 mm3 of Alamar Blue was then added to each well and the plates incubated for another 2 h. Then the plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. The IC50 values were calculated by linear regression [11] from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Cooperation, Sunnyvale, CA, USA). Podophyllotoxin (Sigma P4405) was used as control.
References
Silva JV, Santos SdS, Machini MT, Giarolla J (2021) J Drug Target 29:269
Figarella K (2021) Microb Cell 8:73
Chan-Bacab MJ, Reyes-Estebanez MM, Camacho-Chab JC, Ortega-Morales BO (2021) Molecules 26:1388
Mohsin NUA, Seebacher W, Faist J, Hochegger P, Kaiser M, Mäser P, Belaj F, Saf R, Kretschmer N, Alajlani M, Turek I, Brantner A, Bauer R, Bucar F, Weis R (2018) Eur J Med Chem 143:97
Mohsin NUA, Seebacher W, Hochegger P, Faist J, Saf R, Kaiser M, Mäser P, Weis R (2019) Med Chem Res 28:742
Petritsch M, Seebacher W, Mohsin NUA, Dolensky J, Hochegger P, Kaiser M, Mäser P, Belaj F, Saf R, Kretschmer N, Alajlani M, Brantner A, Bauer R, Schühly W, Weis R (2021) Eur J Med Chem 210:112969
Seebacher W, Faist J, Belaj F, Saf R, Kaiser M, Brun R, Weis R (2015) Monatsh Chem 146:1299
Desjardins RE, Canfield CJ, Haynes JD, Chulay JD (1979) Antimicrob Agents Chemother 16:710
Matile H, Pink JRL (1990) Plasmodium falciparum malaria parasite cultures and their use in immunology. In: Lefkovits I, Pernis B (eds) Immunological methods. Academic Press, San Diego, p 221
Ponnudurai T, Leeuwenberg AD, Meuwissen JH (1981) Trop Geogr Med 33:50
Huber W, Koella JC (1993) Acta Trop 55:257
Baltz T, Baltz D, Giroud C, Crockett J (1985) EMBO J 4:1273
Räz B, Iten M, Grether-Bühler Y, Kaminsky R, Brun R (1997) Acta Trop 68:139
Page C, Page M, Noel C (1993) J Oncol 3:473
Ahmed SA, Gogal RM, Walsh JE (1994) J Immunol Methods 170:211
Acknowledgements
NAWI Graz is acknowledged for supporting the Graz Central Lab Environmental, Plant & Microbial Metabolomics.
Funding
Open access funding provided by University of Graz.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seebacher, W., Kaiser, M., Mäser, P. et al. Benzyl- and dibenzyl tetrahydropyridinylidene ammonium salts with antiplasmodial and antitrypanosomal activity. Monatsh Chem 154, 105–114 (2023). https://doi.org/10.1007/s00706-022-03003-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-03003-w