
Abstract
Zr2(OH)2(XO4)3·4H2O (X = S, Se), Zr(SO4)2·4H2O, and Zr(SeO3)2 were synthesized at low-hydrothermal conditions from mixtures of Zr2O2(CO3)(OH)2, the respective acids, and minor amounts of water. While Zr2(OH)2(XO4)3·4H2O (X = S, Se) and Zr(SO4)2·4H2O form crystals up to several tenths of a mm, Zr(SeO3)2 was mainly obtained as microcrystalline powder, single crystals rarely exceeded 10 μm in size. Samples were investigated by single-crystal X-ray techniques and in the case of Zr(SeO3)2 also by X-ray powder diffraction. The compounds Zr2(OH)2(XO4)3·4H2O (X = S, Se) crystallize in the Ce2(OH)2(SO4)3·4H2O structure type (C2/c, Z = 4, a = 13.034(2) / 13.308(3), b = 6.500(1) / 6.683(2), c = 15.056(3) / 15.383(4) Å, β = 96.27(1) / 96.81(1)°, V = 1267.9(4) / 1358.5(6) Å3 for X = S / Se, respectively). Tetragonal aniprisms Zr[8]O8 are edge-connected to dimers that share corners with XO4 tetrahedra forming a three-dimensional network. Zr(SO4)2·4H2O (Fddd, Z = 8, a = 5.498(1), b = 11.618(3), c = 25.893(6) Å, V = 1653.9(6) Å3) is isotypic with the respective selenate compound. Occasionally, pseudomerohedral twinning is observed, simulating a larger monoclinic C-centered unit cell. Again, tetragonal antiprisms Zr[8]O8 are formed; however, they are corner-linked with SO4 tetrahedra to Zr(SO4)2 layers interconnected solely by hydrogen bonds. Zr(SeO3)2 crystallizes in P21/c, Z = 2; a = 4.9724(3), b = 8.5992(5), c = 6.9447(3) Å, β = 110.128(3)°, V = 278.81(3) Å3 (unit cell from powder data) and belongs to the β-Sn(SeO3)2 structure type established further for Ti(SeO3)2 and Pb(SeO3)2. Isolated ZrO6 octahedra share corners with the selenite groups forming a three-dimensional network.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The presented work is part of long-term investigations on the crystal chemistry of zirconium oxysalts with (XO4)2− or (XO3)2− (X = S, Se) anions synthesized under mild-hydrothermal conditions below 250 °C. So far, the compounds Zr(SeO4)2·H2O and Zr(SeO4)2·4H2O [1], Zr(SeO3)(SeO4), Zr4(SeO3)(SeO4)7, and Zr3(SeO3)(SeO4)5·2H2O [2], as well as M2+Zr(SO4)3 with M = Mg, Mn, Co, Ni, Zn and Cd, and (Fe3+,2+,Zr)2(SO4)3 [3] were obtained and structurally characterized in detail. The present part IV of this series of studies deals with the two isotypic compounds Zr2(OH)2(XO4)3·4H2O (X = S, Se), as well as with Zr(SO4)2·4H2O and Zr(SeO3)2.
The crystal structure of Zr2(OH)2(SO4)3·4H2O was originally described by [4] but the experimental localisation of the hydrogen atoms was not possible at that time. The isotypic analogue Zr2(OH)2(SeO4)3·4H2O was unknown up to now. This structure type is further reported for Ce2(OH)2(SO4)3·4H2O [5].
Zr(SO4)2·4H2O was known since 1956, synthesized and crystallographically characterized by Staritzky and Singer [6]. Later on, [7] determined the crystal structure and refined this compound in space group Fddd but without locating hydrogen atoms. Zr(SO4)2·4H2O is also found in nature as the mineral zircosulfate [8]. It is isotypic with Zr(SeO4)2·4H2O [1] as well as with Hf(SO4)2·4H2O [9]. For Ce(SO4)2·4H2O three modifications were described so far: (i) The Fddd modification—known for decades—but structurally described only in 2007 [10], is isotypic with the title compound and labelled α-form. (ii) The structure of a second, orthorhombic modification (β-form, space group Pnma) was characterized already in 1977 by [11]; it is isotypic with U(SO4)2·4H2O (mineral name behounekite) [12, 13]. Pu(SO4)2·4H2O is dimorph and crystallizes in the α-Fddd and β-Pnma forms [14, 15]. (iii) Finally, [16] reported a third modification crystallizing in space group C2/c. [10] refined both the α- and β-modification of Ce(SO4)2·4H2O and discussed the close structural relations based on a geometrical isomerism. They mentioned pronounced similarities between the α-modification and the crystal structure described in space group C2/c with respect to stereochemistry and topology [10, 11, 16].
For Zr(SeO3)2 no crystal structure was reported so far. Its powder pattern (ICDD entry 50–0336 [17]) was indexed with orthorhombic symmetry (a = 8.568, b = 6.488, c = 15.277 Å); space group Pmmm was proposed based on the absence of systematic extinctions, but several Bragg reflections remained unindexed. Since other M4+(SeO3)2 phases, i.e. the isotypic compounds Ti(SeO3)2 [18], Pb(SeO3)2, and β-Sn(SeO3)2 [19], show similar powder patterns as Zr(SeO3)2 but crystallize in a smaller monoclinic unit cell, we investigated this compound in detail.
Results and discussion
Selected individual and mean bond lengths and angles as well as bond valences ν (calculated according to [20]) of the four title compounds are listed in Table 1. Details about the hydrogen bonds are compiled in Table 2.
Zr2(OH)2(XO4)3·4H2O (X = S, Se)
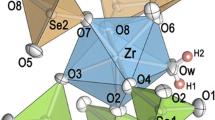
The zirconium atoms in the isotypic compounds Zr2(OH)2(XO4)3·4H2O (X = S, Se) are coordinated by eight oxygen atoms arranged in distorted tetragonal antiprisms. The ligands are represented by the oxygen atoms of two hydroxyl groups (Oh1) and two H2O molecules (Ow8, Ow9), as well as by four oxygen atoms of XO4 tetrahedra (O2, O4, O5, O6). Each two ZrO8 polyhedra are edge-connected to form dimers. The common Oh1–Oh1 edges are shortened to ~ 2.34 Å in both compounds due to repulsion of the two tetravalent central atoms. These dimers are corner-linked with two crystallographically different XO4 tetrahedra (Fig. 1); one has the point symmetry 2, the other is centered on a general position. The oxygen atoms O3 and O7 belong to the XO4 tetrahedra only; furthermore they act as acceptor atoms of three respectively two hydrogen bonds (Table 2). The hydrogen bond lengths D–H···A are ~ 2.7 Å for the H2O molecules but somewhat longer for the hydroxyl groups (2.84 and 2.96 Å for X = S and Se, respectively). Without considering the contributions of hydrogen atoms, the bond valences amount to ~ 1.5 and 1.6 v.u. for the atoms O3 and O7, respectively [20]. The proposed model complies well with calculated bond valences.

Crystal structure of Zr2(OH)2(SO4)3·4H2O in a projection along the crystallographic b-axis
Topologically, the crystal structure is formed by Zr2(OH)2(XO4)3(H2O)4 layers parallel to (101). Eight-membered rings (one corner is concave) and the doubled number of four-membered rings, consisting of alternating Zr atoms and XO4 tetrahedra, build these layers. All types of rings are linked among each other forming rows parallel to [010]. It should be mentioned that two hydrogen bonds, Oh1–Hh1···O3 and Ow8–Hw2···O3, are running within the eight-membered rings of the layers, whereas adjacent layers are interlinked to make up a three-dimensional framework via the Zr2O14 dimers (at their Oh1–Oh1 edge) as well as by Ow8–Hw3···O7, Ow9–Hw4···O3, and Ow9–Hw5···O7 hydrogen bonds.
Zr(SO4)2·4H2O
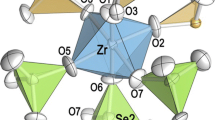
Zr(SO4)2·4H2O (α-modification) [7, and present study] crystallizes in space group Fddd and represents a structure type which further comprises the crystal structures of Zr(SeO4)2·4H2O [1] and of the sulfates of M4+ = Pu, Hf, or Ce [9, 10, 14]. For Zr(SeO4)2·4H2O the presence of a maximum in the residual electron-density (9.7 e Å−3) was observed at the special position 8a (at 1/8 5/8 5/8) [1]; it was attributed to a partially occupied site Zr2 going along with the formation of vacancies at the Zr1 position [8b, at 5/8 5/8 5/8]. The occupation factors for the Zr1 and Zr2 atoms were refined to 0.960(1) and 0.040(1), respectively, indicating a slight site disorder [1]. In the present study on Zr(SO4)2·4H2O, the highest residual electron density amounts 2.2 e Å−3; it again occurs at the 8a position. This slightly enhanced electron density is not considered to be significant in the final refinements of Zr(SO4)2·4H2O. However, a slight disorder cannot be excluded with certainty.
In accordance with an earlier investigation [7], the ZrO8 polyhedra form antiprisms with point symmetry 222, built up by four oxygen atoms of H2O molecules and four O1 atoms. The sulfate groups have point symmetry 2. The ZrO8 polyhedra share four corners—the O1 atoms—with four neighboring sulfate tetrahedra. Vice versa, the sulfate group links between two Zr atoms. Thus eight-membered rings are formed which are linked to Zr(H2O)4(SO4)2 layers running parallel to (001) (Figs. 2, 3a). The remaining two ligands of the sulfate group—atoms O2—act as acceptors of both hydrogen bonds from the H2O molecule (cf. Table 2). The hydrogen bonds Ow–Hw1···O2 are located within the above-mentioned rings, whereas adjacent layers are interconnected only by the hydrogen bond Ow–Hw2···O2. Without the contribution of the hydrogen bonds, the bond valence calculation gives only 1.56 v.u. for the O2 atom.

Crystal structure of Zr(SO4)2·4H2O in a projection along the crystallographic a-axis. The monoclinc cell choice published for ‘γ-Ce(SO4)2·4H2O’ in [16] is shown with dashed lines for comparison. Note: to comply at least approximately with the bond lengths and figures given in [16], the coordinates of O16, O18, H7, H8, H11, and H12 had to be corrected

Single polyhedral sheets in the structures of a Zr(SO4)2·4H2O in a projection onto (001) compared to b ‘monoclinic γ-Ce(SO4)2·4H2O’ according to [16] in a projection onto (\(\overline{2}\) 0 1). Note: to comply at least approximately with the bond lengths and figures given in [16], the coordinates of atoms O16, O18, H7, H8, H11, and H12 had to be corrected
Polyhedral bond-length distortions are quite similar for both compounds Zr(XO4)2·4H2O (X = S, Se) as well as for α-Ce(SO4)2·4H2O [10]. In β-Ce(SO4)2·4H2O (space group Pnma) the respective layers are puckered. For detailed discussions, comparisons, and illustrations of the differences and similarities of these two structure types see [10].
A further modification, i.e. γ-Ce(SO4)2·4H2O, was described in literature [16] in space group C2/c. However, already in Ref. [10] the authors note that “The packing features adopted by the monoclinic form are very similar to the one adopted by the α-form”, and the respective structure layers were compared in their Fig. 3, but not further discussed in detail. A transformation of the unit-cell parameters listed by [16] for the space group C2/c according to [1/3 0 2/3 / 0 1 0 / 1/3 0 –4/3] results in the cell parameters a = 11.988, b = 5.633, c = 26.614 Å, α = 90.00, β = 90.03, γ = 90.00°; they fit with those given by [10] for the orthorhombic modification of α-Ce(SO4)2·4H2O, a = 5.6587(1), b = 12.0469(2), c = 26.7201(3) Å. Applying the same transformation to the atomic coordinates of the two Ce and three S atom positions results in overlapping Ce···Ce distances < 0.009 Å and S···S distances < 0.015 Å. For the O atoms the situation is not that clear as obviously some misprints occur already for the atomic coordinates published in space group C2/c [16]: e.g. O16···O16 (1.21 Å), O13···O18 (2.13 Å), O18···S2 (1.95 Å), although the published H atom positions are conspicuous (see also comments to Figs. 2 and 3). As a consequence, the existence of the structure type of γ-Ce(SO4)2·4H2O must be doubted. Very likely, the phase investigated by [16] actually is the modification α-Ce(SO4)2·4H2O. This close similarity is evident also in our Figs. 2 and 3a, 3b. In the light of the present investigations on the crystal structure of Zr(SO4)2·4H2O with its remarkable twinning behavior (see chapter on single-crystal X-ray diffraction in the experimental part), it seems very likely that crystals of α-Ce(SO4)2·4H2O might be twinned in the same way as described herein, thus simulating a monoclinic C-centered pseudo cell. This assumption is further corroborated by our observation of respective twinning also in Zr(SeO4)2·4H2O, alongside with untwinned single crystals [1].
Zr(SeO3)2
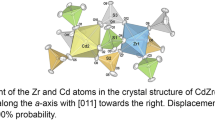
The atomic arrangement in Zr(SeO3)2 consists of isolated ZrO6 octahedra, sharing all corners with trigonal pyramidal selenite groups as illustrated in Fig. 4. Four- and six-membered rings consisting of alternating Zr atoms and selenite groups are formed, linked to tubes running parallel to [100]. The Se atoms of the selenite groups point into these tubes to give space for the lone-pair electrons. The compound is isotypic with Ti(SeO3)2 [18], Pb(SeO3)2, and β-Sn(SeO3)2 [19], a comparison is given in Table 3. It has to be noted that the unit cell volumes of Zr(SeO3)2 and Pb(SeO3)2 are rather similar, while the mean Zr–O bond length resembles more closely that of the SnO6 polyhedron. Actually, based on the single-crystal data, the mean M4+–O bond lengths in this group obey a strict linear correlation with their respective ionic radii. The comparatively high cell volume of Zr(SeO3)2 could be induced by the clearly extended M–O–Se angles, which in turn are negatively correlated with the octahedral bond angle distortion σoct2 (Table 3). As outlined in the section on X-ray powder diffraction, a previous description of Zr(SeO3)2 with orthorhombic symmetry (space group Pmmm) from powder data [17] and the respective ICDD entry 50–0336 have to be revised.

Crystal structure of Zr(SeO3)2 in a projection approximately along the crystallographic a-axis
Analysis of structural similarities
The title compounds and isotypic analogues were geometrically analyzed with the program COMPSTRU as summarized in Table 4 (for details see the experimental section). For the isotypic compounds M2(OH)2(XO4)3·4H2O (M = Zr; X = S, Se [this work]; M = Ce, X = S [5]) the X1 atom (located on a twofold axis, but sharing only two corners with ZrO8-polyhedra) exhibits a larger average shift compared to the X2 atom (on a general position, but three corners shared). However, the X2O4 tetrahedra show a more pronounced rotation. The average shifts of the O atoms belonging to the XO4 tetrahedra in both the Zr compounds amount 0.1520 and 0.1439 Å. Interestingly, these shifts are marginally smaller than the difference between the S/Se–O bond lengths (0.1610 Å). The locations of the H atoms are corroborated, with a displacement between the compounds M = Zr; X = S, Se of only 0.034–0.146 Å. For the structure pairs M = Zr and Ce, X = S, a significant deviation of the Ow positions is evident (0.148 and 0.160 Å). Again, the smaller shift of the S2 atom is notable. In case of the α-M(XO4)2·4H2O structure type (space group Fddd), the smallest degree of lattice distortion and the closest structural similarity are verified between the Zr- and Hf-sulfates. For the selenites M(SeO3)2 (M = Zr, Sn, Ti, Pb) the M atoms are strictly localized due to their point symmetry mmm, but a pronounced shift and rotation of the selenite group is observed. Compared to Zr(SeO3)2, the lattice distortion is large for the Ti-compound but consistently smaller for the Sn- and Pb-representatives; however, the ratios of the corresponding lattice parameters are inverse proportional.
Conclusion
The crystal structures of the oxysalts Zr2(OH)2(XO4)3·4H2O (X = S, Se), Zr(SO4)2·4H2O, and Zr(SeO3)2 were studied by single-crystal X-ray diffraction, Zr(SeO3)2 also by powder techniques. The Zr2(OH)2(XO4)3·4H2O compounds (X = S, Se) are isotypic with Ce2(OH)2(SO4)3·4H2O and crystallize in space group C2/c. Zr(SO4)2·4H2O (space group Fddd) is isotypic with the respective selenate as well as with α-M(SO4)2·4H2O (M = Ce, Pu, Hf). It often shows pseudomerohedral twinning, simulating a monoclinic C-centered cell. By analogy, we postulate that the previously proposed γ-modification of Ce(SO4)2·4H2O (space group C2/c [16]) is most probably identical with α-Ce(SO4)2·4H2O. Finally, Zr(SeO3)2 is monoclinic (space group P21/c) and belongs to the structure type known for Ti(SeO3)2, Pb(SeO3)2, and β-Sn(SeO3)2. A previous indexing of Zr(SeO3)2 with orthorhombic symmetry (space group Pmmm) from powder data [17] and the respective ICDD entry 50-0336 have to be revised.
At the time we started our investigations of zirconium oxysalts [1], more than a dozen of purely inorganic zirconium sulfates, but no selenates or selenites had been structurally described in the literature. In the course of our studies [1,2,3] and present work, we were as yet able to synthesize and characterize seven zirconium sulfates as well as seven zirconium compounds containing [SeO4] and/or [SeO3] anions with interesting stereochemical properties. Currently, in continuation of our research program, the investigation of eight new zirconium oxysalts is in progress. Usually, respective Zr-sulfates and -selenates were found to be isotypic. Besides, the rare element hafnium, substituting zirconium in nature (it is found e.g. as a solid solution with Zr in the mineral zircon, ZrSiO4) is also expected to form isotypic compounds as reported for zirconium. While a series of respective hafnium sulfates are listed in the ICDD database, no hafnium selenates or selenites are evident so far. It is most likely that a thorough study on this class of compounds will be promising, too.
Experimental
Synthesis
The investigated zirconium oxysalts were obtained by low-hydrothermal synthesis from mixtures of Zr2O2(CO3)(OH)2 with concentrated H2SO4, H2SeO4, or H2SeO3, respectively, and a few tenths of a cm3 water filled in Teflon-lined steel vessels of about 5 cm3 volume (filling level ≤ 25%). The autoclaves were heated up to a maximum of 220 °C within several hours, kept at this temperature for one week and finally cooled down slowly to room temperature.
In case of Zr2(OH)2(XO4)3·4H2O (X = S, Se) and Zr(SO4)2·4H2O colorless crystals up to several tenth of a mm formed; they were separated from the remaining liquid, washed in methanol, dried, and embedded in silicone grease. Zr(SeO3)2 was obtained mainly as a very fine-grained white powder, only occasionally crystals with up to 10 µm were obtained; the largest one found was used for the single-crystal X-ray investigation. By variation of reaction conditions (ratio of precursor reagents, temperature, cooling rate, pH value) no significant effect on the grain size of the precipitate could be achieved. In literature, the formation of zirconium selenite only as nanoparticles or microcrystals [21, 22] by reaction of ZrO(NO3)2·H2O and SeO2 at room temperature has been described.
Single-crystal X-ray diffraction data and structure refinement
Selected fragments of Zr2(OH)2(XO4)3·4H2O (X = S, Se), Zr(SO4)2·4H2O, and a tiny chip of Zr(SeO3)2 were prepared for single-crystal X-ray investigations. Data collections were performed on a Bruker APEXII diffractometer equipped with a CCD area detector, an Incoatec Microfocus Source IµS (30 W, multilayer mirror, Mo-Kα), and an Oxford Cryosystems Cryostream 800 Plus LT device; the crystal-detector distance was 40 mm. Zr(SeO3)2 was studied at room temperature, all other samples at 200 K to avoid decomposition and improve the location of the hydrogen atoms. Several sets of phi- and omega-scans with 2° scan-width were combined to achieve respective full-sphere data. For data handling including integration and absorption correction (evaluation of multi-scans) the Bruker Apex3 suite was used [23]. For Zr2(OH)2(XO4)3·4H2O (X = S, Se) the atomic coordinates reported by [4] were taken as starting parameters and the crystal structures were refined by full-matrix least-squares techniques (Shelxl [24]). The positions of the hydrogen atoms were revealed from final difference Fourier maps. Crystal parameters as well as a summary on the data collections and structure refinements are given in Table 5, final atomic positions are compiled in Table 6.
It has to be noted that for Zr(SO4)2·4H2O standard indexing of the ‘single-crystal’ reflex positions led to a monoclinic C-centered unit cell (a = 34.815(5), b = 5.497(1), c = 14.190(3) Å, β = 114.00(1)°, V = 2481.0(8) Å3, Z = 12). This unit cell closely resembles that proposed for γ-Ce(SO4)2·4H2O [16] (a = 35.813, b = 5.633, c = 14.597 Å, β = 113.73°, V = 2695.7 Å3), which is one of three modifications reported for Ce(SO4)2·4H2O in literature [10, 16]. A refinement of Zr(SO4)2·4H2O in space group C2/c using a modified atomic model (see comments in captions to Figs. 2 and 3) published for γ-Ce(SO4)2·4H2O was performed. Moreover, pseudomerohedral twinning was considered (the transformation matrix [1 0 2 / 0 −1 0 / 0 0 −1] gave a = 34.838, b = 5.497, c = 14.190 Å, β = 114.09°); the twin components were 72(1) and 28(1) %, respectively. At a first glance, the refinement gave a reasonable result (R1 = 0.028). However, an evaluation of the refined atomic arrangement (via the ADDSYMM tool implemented in the program PLATON [25]) revealed the presence of “non-space group” translation vectors. A further analysis of the collected data with the program CELL_NOW [26] indicated the tentative presence of a twinned crystal with F-centered orthorhombic unit cell. A transformation of the atom coordinates formerly obtained in space group C2/c by the transformation matrix [1/3 0 2/3 / 0 1 0 / 1/3 0 −4/3] resulted in the hitherto known unit cell and an atomic arrangement based on space-group symmetry Fddd, also known for α-Ce(SO4)2·4H2O [10]. Finally, the observed reflection pattern is best described by an orthorhombic unit cell (a = 5.498, b = 11.618, c = 25.893 Å, Z = 8) and assuming a twinning (twin matrix [1 0 0 / 0 2/3 1/3 / 0 –5/3 2/3]). The conclusive structure refinement, starting with the atomic positions given in [1, 10], is based on a new processing of the measured reflection intensities of both orthorhombic twin components (Bruker APEX3 program suite [23], a reflection file in format HKLF5 from program TWINABS [27]). The orthorhombic crystal-structure model was refined satisfactorily to R1 = 0.027 with twin components of 73(1) and 27(1)%.
For the refinement of Zr(SeO3)2 (see also next section) the unit-cell setting and the atom labelling scheme were chosen in accordance to literature on the isotypic compounds M(SeO3)2 with M = Ti, Sn, and Pb [18, 19].
Furthermore, the title compounds and isopointal analogues were geometrically analyzed with the program COMPSTRU [28, 29]. It allows a comparison between the configurations of crystal-structure pairs by the degree of lattice distortion (S), the maximal displacement between the atomic positions of the paired atoms (dmax), the arithmetic mean of the displacement between the atomic positions (dav), and the similarity as a function of the differences in atomic positions and the ratios of the corresponding lattice parameters (Δ). The results are compiled in Table 4. The atom labels for the structure types under discussion refer to those given in Table 6.
Powder X-ray diffraction work
Microcrystalline powder of Zr(SeO3)2 was studied at ambient conditions on a Bruker D8-Advance Eco diffractometer system, equipped with a CuKα-optimized LynxEye XE-T position sensitive detector with an angular opening of 3.29° 2θ, primary Ni-filter, fixed divergence slit (FDS = 0.3°), 2.5° soller slits for primary and secondary beam path, a fixed anti-scatter knife edge improved by an in-house variable anti-scatter screen and sample φ-spinning (15 min−1). Sample diameter and FDS setting avoided beam spill in the angular range of the first occurring peak at ~ 17° 2θ. Data were collected from 5 to 125° 2θ with ~ 0.01° 2θ step size and an overall measurement time of 200 min (equivalent to an accumulated step counting time of 330 s), thus leading to an Imax of ~ 35,000 counts. The ambient temperature within the Eco-system enclosure was about 30(2) °C. Identification and characterization of crystalline phases was undertaken using the program EVA [30]. Consecutively, the advanced qualitative data treatment based on indexing [31], whole powder pattern decomposition [32], and structure refinement [33] was done with the program TOPAS [34]. The atomic coordinates of the isotypic compounds M(SeO3)2 with M = Ti, Sn, and Pb [18, 19] were used as starting structural parameters.
For the preliminary data evaluation, a Pawley refinement based on the orthorhombic unit cell given in literature [17] was performed, clearly demonstrating a misfit of calculated and observed peak positions (e.g. d-values: 4.09, 3.16, 3.14 Å). The corresponding intensity-unconstrained refinement using the monoclinic cell with space group P21/c revealed a satisfactory fit with Rwp = 7.1% and a GOF of 1.81. The consecutive Rietveld refinement based on the structural model gave a criterion of fit with Rwp = 27.7%, invoking a preferred orientation model along [001] and [012] the refinement converged with Rwp = 10.1%, a value close to the previous Pawley refinement. The refined structural parameters of the Rietveld refinement are given in Table 6, the final Rietveld plot is shown in Fig. 5. X-ray powder-diffraction data are listed in Table 7.

Rietveld plot after the structure refinement of Zr(SeO3)2. Data: observed (black), calculated (red) and difference (gray). The lower tick marks (blue) represent the peak positions of Zr(SeO3)2 (color figure online)
Further details of the crystal-structure investigations may be obtained from the joint CCDC/FIZ Karlsruhe online deposition service: https://www.ccdc.cam.ac.uk/structures/ by quoting the deposition number CSD-2122296 (S), 2122297 (Se) for Zr2(OH)2(XO4)3·4H2O (X = S, Se), 2122298 for Zr(SO4)2·4H2O, and 2122299 for Zr(SeO3)2. An extended Table 6 including anisotropic displacement parameters is available as supplementary information.
References
Giester G, Wildner M (2018) Monatsh Chem 149:1321
Wildner M, Giester G (2019) Monatsh Chem 150:593
Giester G, Talla D, Wildner M (2019) Monatsh Chem 150:1877
McWhan DB, Lundgren G (1966) Inorg Chem 5:284
Lindgren O (1977) Acta Chem Scand A 31:163
Staritzky E, Singer J (1956) Analyt Chem 28:553
Singer J, Cromer DT (1959) Acta Crystallogr 12:719
Kapustin YL (1965) Zap Vses Mineral Obshch 94:530 (in Russian)
Kalaji A, Skanthakumar S, Kanatzidis MG, Mitchell JF, Soderholm L (2014) Inorg Chem 53:6321
Casari BM, Langer V (2007) J Sol State Chem 180:1616
Lindgren O (1977) Acta Chem Scand A 31:453
Plasil J, Fejfarova K, Novak M, Dusek M, Skoda R, Hlousek J, Cejka J, Majzlan J, Sejkora J, Machovic V, Talla D (2011) Mineral Mag 75:2739
Kierkegaard P (1956) Acta Chem Scand 10:599
Wilson RE (2011) Inorg Chem 50:5663
Jayadevan NC, Singh Mudher KD, Chackraburtty DM (1982) Z Kristallogr 161:7
Filipenko OS, Leonova LS, Atovmyan LO, Shilov GV (1998) Dok Akad Nauk 360:73 (in Russian)
ICDD, International Centre for Diffraction Data, PDF 50–0336
Legros JP, Galy J (1978) Comp Rend Hebd. Ser C Sci Chim 286:705
Steinhauser G, Luef C, Wildner M, Giester G (2006) J Alloys Comp 419:45
Brese NE, O’Keeffe M (1991) Acta Crystallogr B 47:192
Henry J, Mohanraj K, Kannan S, Barathan S, Sivakumar G (2013) J Chil Chem Soc 58:1759
Naik CC, Salker AV (2018) Mater Res Express 5:045023
Bruker (2020) Apex3 suite. Bruker AXS, Karlsruhe, Germany
Sheldrick GM (2015) Acta Crystallogr C 71:3
Spek AL (2003) J Appl Cryst 36:7
Sheldrick GM (2003) CELL_NOW, University of Göttingen
Sheldrick GM (2012) TWINABS 2012/1. Bruker, Madison, Wisconsin, USA
de la Flor G, Orobengoa D, Tasci E, Perez-Mato JM, Aroyo MI (2016) J Appl Cryst 49:653
Bergerhoff G, Berndt M, Brandenburg K, Degen T (1999) Acta Crystallogr B 55:147
Bruker (2019) DIFFRAC.SUITE EVA, version 5.1. Bruker AXS, Karlsruhe, Germany
Coelho AA (2003) J Appl Cryst 36:86
Pawley GS (1981) J Appl Cryst 14:357
Rietveld HM (1969) J Appl Cryst 2:65
Bruker (2017) DIFFRAC.SUITE TOPAS, version 6. Bruker AXS, Karlsruhe, Germany
Acknowledgements
This study was financially supported by University Vienna grants IS526001 and IP532010. We are grateful to three anonymous reviewers for their helpful comments.
Funding
Open access funding provided by University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wildner, M., Lengauer, C.L., Effenberger, H. et al. Contributions to the stereochemistry of zirconium oxysalts—part IV: syntheses and crystal structures of Zr2(OH)2(XO4)3·4H2O (X = S, Se), Zr(SO4)2·4H2O, and Zr(SeO3)2. Monatsh Chem 153, 139–151 (2022). https://doi.org/10.1007/s00706-021-02887-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-021-02887-4